Le scRNA-seq : un outil essentiel pour l’exploration de la mégacaryopoïèse – Application à la thrombopénie liée au facteur de transcription ETV6
La mégacaryopoïèse et la formation des plaquettes
Les mégacaryocytes (MK) se différencient à partir de la cellule souche hématopoïétique (CSH) en trois étapes
(Figure 1). La première étape est une phase classique de prolifération des progéniteurs (1). Elle est suivie par une phase spécifique aux MK, caractérisée par une polyploïdisation du noyau par endomitoses. Enfin, le MK subit une maturation cytoplasmique avec une synthèse accrue d’ARNm et de protéines, et la biogenèse des organelles plaquettaires, notamment les granules α, qui stockent des protéines clés. À l’issue de cette étape, les MK migrent de la niche ostéoblastique vers la niche vasculaire pour accomplir leur principale fonction : la libération des plaquettes. Le processus de différenciation commence avec les CSH multipotentes, qui perdent progressivement leur capacité d’auto-renouvellement pour devenir des progéniteurs bipotents mégacaryocytaires/érythroïdes (MEP), pouvant se différencier en MK ou en cellules érythroïdes. Les MEP, à travers des étapes finement régulées, donneront des progéniteurs mégacaryocytaires qui se différencieront en MK immatures, puis matures. Les MK immatures peuvent mesurer jusqu’à 30 µm avec un rapport noyau/cytoplasme élevé, tandis que dans leur état mature, ils peuvent atteindre jusqu’à 160 µm. La ploïdie modale d’un MK mature est de 16N, mais elle peut atteindre 64 voire 128N. La grande quantité d’ADN dans chaque cellule contribue à une forte concentration d’ARN au moment de la formation des proplaquettes. La régulation de la mégacaryopoïèse implique l’action concertée d’un certain nombre de facteurs parmi lesquels des cytokines et des facteurs de croissance, dont la thrombopoïétine qui joue un rôle majeur, ainsi qu’un certain nombre de facteurs de transcription (FT) : RUNX1, FLI1, GATA1, ETV6… Un défaut à un stade de ce processus peut entraîner une production anormale de plaquettes et contribuer au développement de pathologies plaquettaires constitutionnelles (PPC).
Les PPC forment un groupe hétérogène de maladies, tant en termes de symptômes, de gravité que d’évolution, affectant à la fois la formation et/ou la fonction des plaquettes. Ces pathologies reflètent la complexité des mécanismes régulant la mégacaryopoïèse et peuvent servir de modèle pour l’étude de cette dernière. Les patients atteints présentent généralement des symptômes hémorragiques de type cutanéo-muqueux (épistaxis, purpura, saignements gingivaux, ménorragies…), mais certains phénotypes incluent aussi des atteintes extra-hématologiques ou un risque accru de cancers. L’expression clinique est très variable, allant de formes bénignes à des manifestations potentiellement fatales. Grâce aux progrès des techniques de séquençage génétique, plus de 40 gènes impliqués dans les thrombopénies ont pu être identifiés. Ces variants touchent principalement des gènes liés à la différenciation des MK, à l’organisation du cytosquelette, ou à l’activité de FT (2).
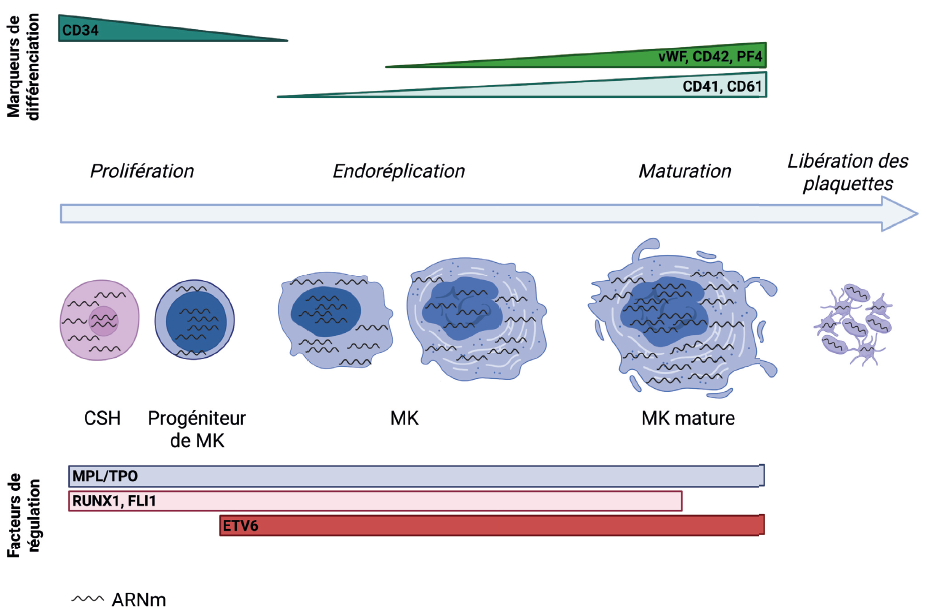
Figure 1 : Aperçu de la mégacaryopoïèse.
Figure 1: Overview of Megakaryopoiesis.
La mégacaryopoïèse se déroule en trois grandes étapes : la prolifération des cellules dans les premiers stades, l’endoréplication (ou endomitose) conduisant à la formation de mégacaryocytes (MK) polyploïdes, puis la phase de maturation cytoplasmique. Cette dernière est marquée par une augmentation de la synthèse d’ARN messagers et de protéines, ainsi que par la biogenèse des organelles plaquettaires, notamment les granules α, qui stockent des protéines clés. Finalement, les MK produisent des proplaquettes, précurseurs des plaquettes circulantes. L’analyse des antigènes de surface a permis de défi nir les stades de diff érenciation cellulaire. Les cellules progénitrices expriment CD34, tandis que les cellules plus diff érenciées présentent des marqueurs plus spécifi ques comme le complexe α2b β3 (GPIIb/CD41 et GPIIIa/CD61), GPIX (CD42a), GPV (CD42d), GPIb (CD42b), le facteur von Willebrand (vWF) et le facteur plaquettaire 4 (PF4). La régulation de la mégacaryopoïèse repose sur l’action coordonnée de cytokines et de facteurs de croissance, notamment la thrombopoïétine (TPO) et son récepteur MPL, ainsi que sur l’activité de facteurs de transcription tels que RUNX1, FLI1 et ETV6 indispensables à la diff érenciation et à la maturation des MK.
Modèle in vitro
En raison des difficultés techniques liées à l’extraction des MK de la moelle osseuse et du faible rendement obtenu, les modèles in vitro ont pris une place essentielle dans l’étude de la mégacaryopoïèse et des PPC. Les MK peuvent être dérivés de cellules souches pluripotentes induites (iPSC) par l’ajout de cytokines ou la surexpression de FT, ou à partir de lignées cellulaires, comme les HEL (dérivées d’un patient atteint d’érythroleucémie), les Meg-01 (issues d’une leucémie mégacaryoblastique chronique) ou la lignée imMKCL (immortalisée à partir d’iPSCs d’un individu sain) (3). Le modèle le plus couramment utilisé reste la différenciation de cellules souches/progénitrices CD34+ (issues de la moelle osseuse, du sang périphérique ou du sang de cordon), cultivées avec un cocktail de cytokines. Ce système s’est révélé particulièrement utile pour caractériser les PPC, étudier de nouveaux mécanismes de différenciation mégacaryocytaire, et tester l’effet de pathogènes ou d’agents thérapeutiques. Toutefois, les populations cellulaires obtenues par cette méthode, y compris dans les cultures témoins, restent encore insuffisamment caractérisées.
Le séquençage des transcrits à l’échelle unicellulaire (ou single-cell RNA-seq, scRNA-seq)
Le transcriptome désigne l’ensemble des transcrits présents dans une cellule à un instant donné, reflétant les gènes exprimés. Contrairement au génome, globalement stable, le transcriptome est dynamique. Il évolue en fonction des signaux internes et externes, traduisant ainsi l’état fonctionnel, ou phénotype, de la cellule. L’analyse du transcriptome permet de comprendre la fonction des gènes, leur régulation et leur impact dans l’émergence de maladies. Avec l’avènement du séquençage à haut débit, les approches transcriptomiques ont considérablement évolué, passant des analyses ciblées (comme les puces à ADN) à l’étude globale du transcriptome (RNA-seq). Toutefois, les méthodes de RNA-seq dites « en vrac » (bulk RNA-seq) reposent sur l’hypothèse d’une homogénéité cellulaire et masquent l’hétérogénéité fonctionnelle entre cellules, y compris au sein de populations triées. Le séquençage à l’échelle unicellulaire (scRNA-seq) permet désormais d’analyser simultanément le transcriptome de milliers de cellules, de façon non supervisée (sans a priori), c’est-à-dire sans hypothèse préalable sur l’identité ou les fonctions des sous-populations, révélant ainsi la diversité et l’hétérogénéité cellulaires d’un échantillon. Les techniques de scRNA-seq se divisent en deux catégories : celles basées sur des plaques, isolant les cellules dans des puits, et celles microfluidiques, utilisant des microgouttelettes aqueuses dans un milieu hydrophobe.
Les étapes essentielles d’un scRNA-seq en gouttelettes
Cette revue se concentre sur le système microfluidique Chromium, développé par 10X Genomics, qui est aujourd’hui l’un des outils les plus largement utilisés pour le scRNA-seq (4). La première étape consiste à obtenir une suspension de cellules individuelles. Pour les cellules déjà en suspension, aucune préparation particulière n’est nécessaire, tandis que les cellules adhérentes ou extraites de tissus solides doivent être détachées mécaniquement ou par méthode enzymatique, tout en veillant à préserver leur intégrité. Les cellules sont ensuite encapsulées individuellement dans des gouttelettes d’émulsion (Figure 2A). Pour garantir une résolution à cellule unique, la suspension cellulaire doit être faiblement concentrée, avec une dilution limitante assurant l’absence de cellule dans la majorité des gouttelettes, tandis que les autres en contiennent le plus souvent une seule. Chaque gouttelette contient les réactifs de rétrotranscription et une bille appelée GEM (Gel bead in Emulsion) recouverte de dizaines de milliers de code-barres d’oligonucléotides. Après encapsulation, les cellules sont lysées et leurs ARN messagers sont capturés, puis rétrotranscrits en ADNc avec l’ajout d’identifiants uniques permettant de relier chaque transcrit à sa cellule d’origine (Figure 2B). Les ADNc sont ensuite amplifiés et convertis en une librairie indexée, prête pour le séquençage à haut débit (Figure 2C). Cette étape marque la fin de la préparation expérimentale et constitue le point de départ de l’analyse bio-informatique.
Avantages et Limites
Le scRNA-seq permet de détecter des sous-populations cellulaires hétérogènes et d’approfondir la compréhension de mécanismes biologiques complexes. Cependant, cette méthode présente certaines limites, notamment une sensibilité parfois réduite pour les gènes faiblement exprimés et une résolution limitée pour l’analyse des longs transcrits. De plus, étant exclusivement transcriptomique, elle ne permet pas de capturer l’ensemble des phénomènes moléculaires influençant l’identité cellulaire, sa fonction, et ses réponses aux stimuli, tels que les modifications post-transcriptionnelles, la régulation épigénétique ou les interactions avec le microenvironnement.
L’analyse bio-informatique des données scRNA-seq
L’analyse bio-informatique des données de scRNA-seq se déroule en trois étapes principales : l’étape primaire (ou de prétraitement), l’étape secondaire (ou d’analyse exploratoire), et l’étape d’interprétation biologique. Chacune repose sur des outils spécifiques et une interaction étroite entre bio-informaticiens et biologistes.
Analyse primaire – Du séquençage aux matrices de comptes
L’étape primaire transforme les lectures brutes de séquençage (fichiers FASTQ) en matrices de comptes, résumant l’expression de chaque gène dans chaque cellule, généralement via le pipeline CellRanger (10x Genomics). Cette étape génère un rapport de qualité, essentiel pour évaluer la fiabilité des données (Tableau 1). Plusieurs métriques clés doivent être examinées : le nombre estimé de cellules, qui reflète le rendement de l’expérience ; le nombre médian de gènes exprimés et UMI par cellule, indicateurs de la complexité transcriptionnelle et de la profondeur de séquençage ; le pourcentage de lectures alignées au génome de référence, ainsi que la proportion d’UMI associés à des gènes, permettant d’estimer la qualité de l’alignement et le niveau de bruit technique (Tableau 1). Un faible rendement cellulaire peut indiquer un problème de capture, notamment pour les types cellulaires volumineux comme les MK. Bien que le système Chromium tolère théoriquement des cellules jusqu’à 65 µm, il est optimisé pour des tailles inférieures à 30 µm. Dans nos études, les MK ont donc été analysés avant leur maturation complète afin d’optimiser leur récupération. Pour des cellules plus grandes, un protocole spécial d’isolement des noyaux peut être utilisé pour contourner les limites physiques du système. La saturation du séquençage est une autre métrique clé qui permet d’évaluer si une couverture supplémentaire est nécessaire pour capturer toute la diversité transcriptionnelle de l’échantillon. Le Barcode Rank Plot, qui classe les codes-barres cellulaires en fonction de leur abondance en UMI, aide à distinguer les cellules viables du bruit de fond. Un graphique en forme de genou (knee plot) présente une chute brutale, marquant la frontière entre les cellules intactes et le bruit de fond, constitué d’ARN ambiant et de cellules partiellement dégradées. En l’absence de transition nette, une contamination ou un faible rendement de capture peut être suspecté.
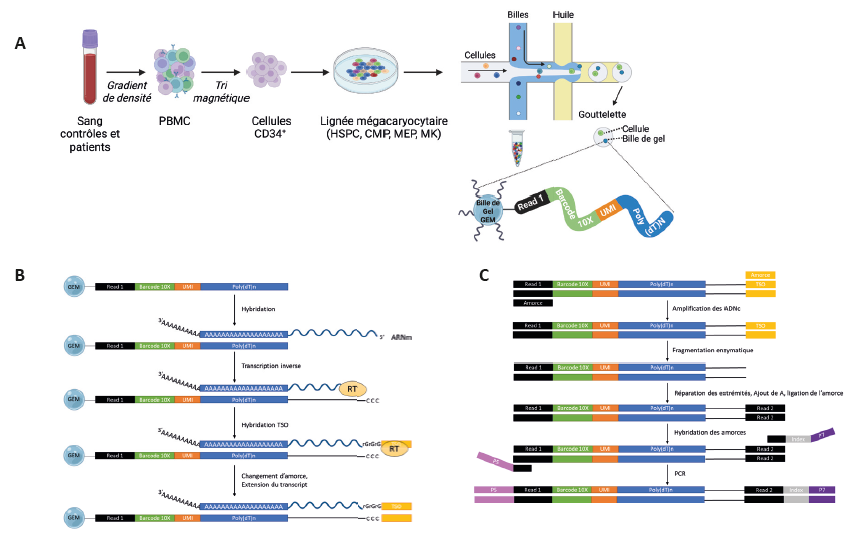
Figure 2 : Protocole de scRNA-seq 10X Genomics basé sur l’isolement par gouttelette.
Figure 2: Workfl ow of the 10X Genomics scRNA-seq Protocol Using Droplet-Based Isolation.
Des cellules CD34+ périphériques ont été isolées par gradient de densité et tri magnétique. Ces cellules ont été diff érenciées en MK dans
un milieu sans sérum (StemSpan™ SFEM II) enrichi en cytokines spécifiques (SCF, IL6, TPO, IL9).
A – Isolement des cellules
Adapté de 10x Genomics. La microfluidique est une technologie qui permet de manipuler des fluides dans des microcanaux. Grâce à
cette technique, chaque cellule est isolée dans une gouttelette d’huile contenant des réactifs (polymérase, oligonucléotide, transcriptase
inverse, etc.) et une bille spécifique appelée GEM (Gel Bead in Emulsion). Chaque GEM est recouverte de dizaine de milliers de code-barres d’oligonucléotides comprenant une amorce PCR (Read 1), un code-barres 10x (16bp), qui constitue un identifiant unique pour chaque bille, et donc pour chaque cellule (10x Genomics propose environ 750 000 codes-barres), ainsi que des identifiants moléculaires uniques (UMI Unique Molecular Identifiers), qui sont des courtes séquences aléatoires uniques à chaque fragment associé à la bille. Il y a donc plusieurs UMI par bille. Cet identifiant est crucial pour éviter les biais d’amplification. Si une séquence est accidentellement trop amplifiée dans une gouttelette, elle pourra être détectée, car le même UMI sera représenté plusieurs fois. Enfin, la séquence polyT permet la fixation des ARN messagers (ARNm) par complémentarité avec leurs queues polyA.
B – Rétrotranscription et ajout de code-barres à l’intérieur de chaque gouttelette
Après isolement dans les gouttelettes, les cellules sont lysées par chauff age et les ARNm libérés sont capturés à la surface de la GEM par
leurs queues polyA. Les nouvelles séquences code-barres + UMI + ARNm sont alors converties en ADN complémentaires (ADNc). Tout
d’abord, un oligonucléotide s’hybride à la molécule d’ARN et la rétrotranscriptase (RT) commence à polymériser. La rétrotranscriptase
utilisée a une activité terminale transférase rajoutant des cytosines sur le brin d’ADNc. Cette terminaison permet l’hybridation d’une
séquence appelée « template switch oligo » (TSO) en 5’ du transcrit, complétée en 3’ du brin d’ADNc. La partie 3’ du transcrit est également complétée par complémentarité avec la région 5’ du brin d’ADNc.
C – Construction de la librairie ADNc
Après incubation des gouttelettes, des ADNc double brin sont produits, contenant les identifiants uniques de cellule et de molécule. L’huile est ensuite retirée, et les ADNc sont regroupés dans une solution débarrassée des réactifs et séquences libres. Les ADNc sont amplifiés par PCR à l’aide des amorces Read 1 et TSO. La taille des amplicons est optimisée par fragmentation enzymatique, puis des séquences « Read 2 » et des index pour le multiplexage sont ajoutés, générant une librairie compatible avec les plateformes de séquençage pairedend Illumina. La lecture du transcrit s’effectue sur 90 nucléotides à partir de son extrémité 3’.
Tableau 1 : Comprendre les rapports Cell Ranger de 10X Genomics (cellules analysées au J6 de la culture).
Table 1: Understanding Cell Ranger reports from 10X Genomics (cells analysed on day 6 of culture).

Analyse secondaire, des matrices de comptes au clustering
L’analyse secondaire applique des méthodes statistiques pour filtrer les artefacts, normaliser les données, réduire leur dimensionnalité et regrouper les cellules en sous-populations homogènes selon leurs profils transcriptionnels. Contrairement à l’analyse primaire, qui vérifie les données brutes, cette étape intègre des critères de contrôle qualité plus détaillés. Les cellules sont filtrées en fonction du nombre de gènes et de transcrits détectés pour éliminer celles de mauvaise qualité, telles que les vides ou les multiplets. Les seuils de filtrage (300-6000 gènes, 500-25000 transcrits) doivent être adaptés aux lignées hématopoïétiques, afin de préserver les cellules présentant un contenu ARN atypique (plaquettes, MK), qui risqueraient d’être exclues à tort avec des seuils standards. Un taux élevé de gènes mitochondriaux peut indiquer des cellules apoptotiques ou lysées. Les 2 000 gènes les plus variables sont sélectionnés pour refléter les variations biologiques essentielles. Une régression du cycle cellulaire est effectuée pour éliminer les biais liés à la prolifération. La réduction de dimension simplifie ensuite les données complexes en les projetant dans un espace de plus faible dimension (souvent 2 ou 3 dimensions), tout en préservant les relations importantes entre les cellules. Cette étape est suivie d’un clustering permettant d’identifier des sous-populations homogènes, visualisées par des méthodes comme l’UMAP (Uniform Manifold Approximation and Projection).
Interprétation biologique
L’annotation des types cellulaires consiste à identifier les différents types de cellules présents dans un échantillon, en utilisant des marqueurs de référence (gènes spécifiques à chaque type cellulaire) et les différences d’expression entre les groupes de cellules (appelées clusters). Cette étape est cruciale car une mauvaise identification peut fausser toute l’interprétation des résultats. Parfois, il peut être nécessaire de réaliser un sous-clustering, c’est-à-dire de diviser les clusters en sous-groupes plus petits, pour mieux distinguer des populations de cellules qui se ressemblent beaucoup, mais qui sont en réalité différentes. Cela peut être particulièrement utile lorsque des cellules matures et immatures coexistent dans un même cluster, une situation souvent difficile à distinguer lors du premier niveau de clustering. Une fois les types cellulaires identifiés, on procède à l’analyse des gènes différentiellement exprimés (DEG), c’est-à-dire les gènes dont l’expression varie significativement entre différents clusters de cellules – par exemple, entre un groupe témoin et un groupe de patients. Ces gènes sont ensuite soumis à une analyse d’enrichissement fonctionnel qui vise à donner du sens biologique à cette liste de gènes : sont-ils impliqués dans la division cellulaire, dans la réponse immunitaire, dans la synthèse de protéines, etc. ? Pour cela, la liste des gènes est comparée à des bases de données spécialisées comme GO (Gene Ontology), qui classe les gènes selon leur fonction, localisation cellulaire et implication dans divers processus, ou encore KEGG (Kyoto Encyclopedia of Genes and Genomes), qui regroupe les gènes selon les grandes voies métaboliques ou de signalisation auxquelles ils participent. L’objectif est de repérer certains processus surreprésentés dans la liste de gènes. Cela suggère qu’ils sont probablement activés ou perturbés dans le groupe de cellules étudié.
En complément de cette analyse « statique » des types cellulaires, on peut également étudier leur trajectoire d’évolution au cours du temps, notamment lors de leur différenciation. Cette analyse repose sur la reconstruction d’un pseudo-temps, une sorte de temps virtuel qui permet d’ordonner les cellules selon leur état de maturation, sans pour autant refléter la durée exacte de chaque étape. Cela permet de visualiser comment les cellules évoluent d’un état à un autre au sein d’un processus dynamique. Par exemple, l’expression retardée de marqueurs tels que GP9 et PF4 chez les patients porteurs de variants ETV6 suggère des altérations dans la maturation mégacaryocytaire.
Enfin, l’analyse des réseaux de régulation transcriptionnelle, à l’aide d’outils comme SCENIC ou iCistarget, permet de comprendre quels FT contrôlent les changements observés dans les cellules. Cela aide à reconstituer les circuits de régulation moléculaire impliqués dans leur évolution.
Limites
L’annotation des types cellulaires repose sur des marqueurs qui ne sont pas toujours parfaitement spécifiques d’un type cellulaire, ce qui peut rendre l’attribution des cellules parfois ambiguë. De plus, certains gènes sont partagés entre plusieurs types cellulaires, compliquant ainsi la classification. L’analyse d’enrichissement fonctionnel peut parfois produire des résultats redondants ou imprécis, notamment lorsque des gènes similaires apparaissent dans différentes voies biologiques, ce qui nécessite une analyse de synthèse pour éliminer les voies redondantes et extraire les informations pertinentes. Enfin, bien que le pseudo-temps soit utile pour voir comment les cellules évoluent, il ne doit pas être confondu avec un temps biologique réel, car il s’agit d’une évaluation relative de l’ordre des états cellulaires. Quant aux outils de reconstruction des réseaux de régulation, ils identifient les FT actifs dans un groupe de cellules donné, mais ces outils ne tiennent pas compte des régulations négatives (celles qui inhibent l’expression des gènes), ce qui peut omettre certains mécanismes essentiels.
Le séquençage de plusieurs échantillons
La comparaison de plusieurs échantillons est essentielle dans la plupart des applications de scRNA-seq, mais elle présente des défis, notamment en termes de réplicats biologiques et d’effets « batch ». L’effet « batch » fait référence à la variabilité observée entre les échantillons qui n’est pas liée à des différences biologiques, mais à des facteurs techniques d’expérimentation, tels que les différences de traitement, de réactifs ou de plateformes utilisées. Cette variabilité peut masquer ou imiter des différences d’expression génique, compromettant ainsi l’interprétation des résultats. Augmenter le nombre de réplicats est crucial pour garantir la reproductibilité, mais cette approche peut être limitée par les coûts élevés et les effets « batch » potentiels. Pour les atténuer, plusieurs méthodes d’harmonisation des données peuvent être mises en œuvre. Des outils bio-informatiques comme Seurat IntegrateData ou Harmony permettent d’aligner les sous-populations cellulaires en corrigeant les variations non biologiques entre échantillons, assurant ainsi un clustering plus cohérent. Une autre stratégie consiste à utiliser des protocoles de multiplexage tels que le cell hashing, permettant de regrouper plusieurs échantillons dans une même librairie. Ce procédé consiste à marquer les cellules avec des anticorps dirigés contre des protéines de surface ubiquitaires, telles que la β-2 microglobuline. Ces anticorps sont préalablement marqués à la streptavidine, ce qui leur permet de se lier à l’extrémité 5’ d’un code-barres d’oligonucléotides biotinylés via un pont disulfure réductible. Ces code-barres, appelés HTO (pour hashtag oligonucléotides), contiennent une séquence d’identification unique et une amorce PCR. Après encapsulation des cellules, les code-barres sont libérés, amplifiés et préparés pour le séquençage Illumina. L’analyse bio-informatique permet de quantifier les HTO et de retrouver l’échantillon d’origine pour chaque cellule.
Application du scRNA-seq à l’étude de la mégacaryopoïèse in vitro : cas de la thrombopénie liée aux variants d’ETV6
Les variants constitutionnels du gène ETV6, un FT clé dans l’hématopoïèse, sont responsables d’une thrombopénie et d’un risque accru de développer une hémopathie maligne. Bien que les études menées jusqu’à présent aient fourni des informations importantes sur le rôle joué par ETV6 dans la mégacaryopoïèse et la formation des plaquettes, les mécanismes moléculaires qui sous-tendent cette maladie ne sont pas connus (5). Un défi majeur dans l’étude des thrombopénies liées à ETV6 est l’absence de modèles murins fiables et entièrement représentatifs de la pathologie humaine. Cependant, des modèles in vitro, largement utilisés et basés sur la différenciation de MK à partir de cellules CD34+ de patients, reproduisent des caractéristiques clés de la thrombopénie associée aux variants d’ETV6, telles que le défaut de polyploïdisation et de formation des proplaquettes (5). Bien que facilement mis en œuvre, ce modèle nécessite une caractérisation transcriptionnelle plus fine pour mieux comprendre la dynamique de la différenciation. Notre objectif était de comprendre les mécanismes transcriptionnels liant les variants d’ETV6 à la thrombopénie et à la prédisposition aux hémopathies malignes, tout en caractérisant la mégacaryopoïèse physiologique dans ce modèle (6). Les analyses scRNA-seq ont été réalisées sur des cellules CD34+ issues de deux témoins sains et deux patients porteurs de variants ETV6 différents, cultivées dans un milieu favorisant la différenciation mégacaryocytaire, au jour 6 et au jour 11 (Figure 2A). L’utilisation du multiplexage par HTO a permis d’analyser simultanément les quatre échantillons.
Analyse pas à pas
La réduction dimensionnelle appliquée aux cellules témoins a permis de visualiser les variations transcriptionnelles survenant après 6 et 11 jours de culture. Cette analyse a révélé l’activation de programmes géniques distincts à chaque étape de la différenciation (Figure 3A). Cette approche projette chaque cellule dans un espace bidimensionnel, où elle est représentée par un point. De manière simplifiée, plus les points sont proches les uns des autres, plus les profils transcriptomiques des cellules correspondantes sont similaires.
Le clustering a ensuite permis de regrouper les cellules selon la similarité de leur expression génique. Le nombre final de clusters dépend du paramètre de résolution défini lors de l’analyse. Dans ce cas, une résolution élevée a été choisie afin de favoriser la détection de sous-populations de petite taille. Quinze clusters ont ainsi été identifiés et numérotés par ordre croissant en fonction du nombre de cellules qu’ils contiennent (Figure 3B).
L’analyse combinée, à la fois supervisée et non supervisée, des clusters chez les témoins a permis de caractériser les types cellulaires présents dans les cultures. L’approche supervisée repose sur l’analyse de l’expression de gènes marqueurs spécifiques des principaux types cellulaires attendus dans ce système de différenciation : cellules souches/progénitrices hématopoïétiques (HSPC), progéniteurs myéloïdes communs (CMP), progéniteurs granulocytaires et macrophagiques (GMP), MEP et MK (Figure 3C). Ces gènes ont été sélectionnés grâce à une analyse approfondie de la littérature. L’analyse non supervisée, quant à elle, repose sur l’identification des gènes les plus fortement exprimés au sein de chaque cluster, indépendamment des connaissances a priori (diagramme en points non montré). La combinaison de ces deux approches a permis d’identifier et de valider la présence des principales populations hématopoïétiques au sein de la culture. Le cluster 10 a été identifié comme HSPC en raison de l’expression de gènes spécifiques tels que PROM1, CRHBP, FLT3, HOPX et AVP. Les clusters 11 et 7 ont été associés aux CMP, via l’expression de CPA3, PRG2, CLC et TPSAB1. Cependant, le cluster 11, exprimant également des gènes associés aux MK (VWA5A, LMNA, CAVIN2, LAT, CD9, ITGA2B), a été défini comme des CMP prédisposés aux MK. Ce cluster nécessite des investigations fonctionnelles supplémentaires. Seul un tri cellulaire suivi d’une culture in vitro permettrait de déterminer avec certitude le devenir de ces cellules. Le cluster 13 a été désigné GMP en raison de l’expression des gènes MPO, ELANE et PRTN3. Les clusters 9, 5, 4, 0 et 2 ont été classés comme MEP : le cluster 9 correspond à des MEP précoces (early MEP); le cluster 5 a des MEP-érythroïde (expression des gènes érythrocytaires HBB, TFR2 et ANK1) et les clusters 4, 0 et 2 ont été classés comme MEP-MK en raison de l’expression de gènes MK (GP9, GP6, GP1BA, MPIG6B) et de l’augmentation progressive de MYH9. Les clusters 1, 6, 3, 8 et 12, majoritairement présents à J11, ont été définis comme MK (GP6, GP9, PF4, P2RY1), avec une progression de l’immaturité (cluster 1) à la maturité (cluster 12). Enfin, le cluster 14, principalement observé au jour 11, a été désigné comme des particules semblables à des plaquettes (PLP pour platelet-like particles). Il se distingue par un contenu en ARN très faible, suggérant une activité transcriptionnelle réduite, mais présente une forte expression de gènes associés aux fonctions plaquettaires et à la biogenèse des granules, tels que STXBP5, RAB27B, GP6, RGS18 et ITGA2B.
Application des signatures transcriptomiques aux cellules des patients porteurs de variants ETV6. L’analyse combinée des approches supervisée et non supervisée des cellules témoins a permis de définir les signatures transcriptomiques des différents types cellulaires présents dans le modèle de culture (Figure 3D-E). Les signatures ont ensuite été appliquées aux cellules des patients. Les mêmes types cellulaires ont été retrouvés (à l’exception du cluster PLP), bien que les proportions soient altérées. Les patients présentaient un enrichissement en progéniteurs immatures et une diminution des MK matures, indiquant un blocage partiel de la différenciation, cohérent avec le phénotype observé (Figure 3F).
Analyse différentielle et voies dérégulées
L’analyse combinée des données des patients et des témoins a permis une comparaison fine des profils transcriptomiques. Les premières divergences majeures sont apparues dès le stade MEP, avec des DEG enrichis dans des voies de la traduction et de la réparation de l’ADN, selon plusieurs bases de données d’annotation fonctionnelle (GO, Reactome, KEGG). Ces dérégulations sont actuellement en cours d’exploration pour mieux comprendre les mécanismes moléculaires responsables de la thrombopénie liée aux variants d’ETV6.
Mise à disposition du pipeline
Afin de favoriser la reproductibilité, nous mettons à disposition un pipeline d’analyse modulable et standardisé sur GitHub : https://github.com/poggiteam/ETV6_2020.
Conclusion et perspectives
L’utilisation du scRNA-seq, couplée à des outils bio-informatiques avancés, constitue une approche puissante pour mieux comprendre les mécanismes moléculaires à l’origine des thrombopénies constitutionnelles et ouvre la voie à de nouvelles stratégies thérapeutiques ciblées. Dans notre étude, cette technologie a permis de caractériser de manière fine un modèle in vitro de différenciation de cellules CD34+ en MK, tout en apportant un nouvel éclairage sur les anomalies transcriptionnelles associées aux variants ETV6. Des altérations significatives ont été observées, notamment dans les voies de la réparation de l’ADN, ce qui est en accord avec le risque accru de leucémies chez ces patients, ainsi que dans la traduction, suggérant de nouveaux axes de recherche (Figure 3G). Les prochaines étapes viseront à explorer le rôle de la traduction dans la mégacaryopoïèse, à travers l’étude de la protéine ribosomale RPS6, avec pour objectif l’identification de cibles thérapeutiques susceptibles de restaurer efficacement la production plaquettaire. Ces travaux auront des applications au-delà du spectre des anomalies ETV6 et seront également précieux pour la biologie fondamentale.
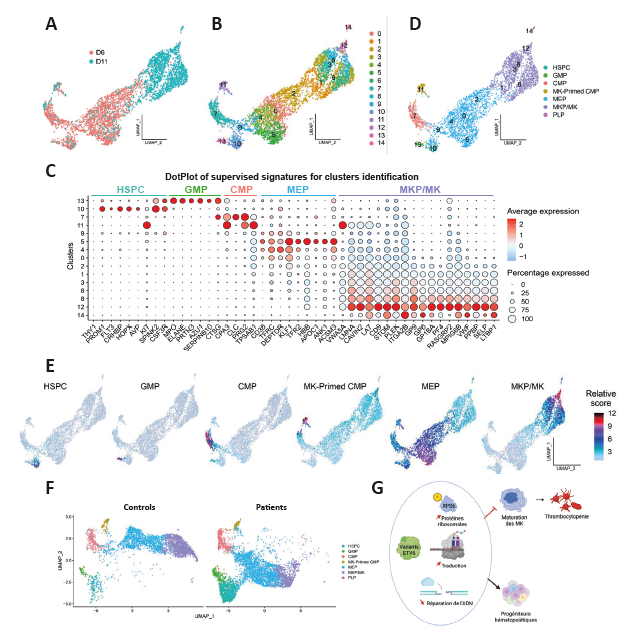
Figure 3 : Analyse par scRNAseq des cellules à diff érents stades de la mégacaryopoïèse.
Figure 3: scRNA-seq Analysis of Cells across Different Stages of Megakaryopoiesis.
Les cellules CD34+ périphériques ont été isolées par gradient de densité et tri magnétique, puis diff érenciées en MK dans un milieu sans sérum (StemSpan™ SFEM II) enrichi en cytokines spécifiques
(SCF, IL6, TPO, IL9). Afi n de combiner plusieurs échantillons (n = 2 témoins sains, n = 2 patients), les cellules ont été marquées avec des anticorps couplés à des codes-barres HTO. Les analyses transcriptomiques ont été réalisées aux jours 6 et 11 en utilisant la technologie 10x Genomics (6). Adaptée de Bigot et coll., J Thromb Haemost, 2023.
A – UMAP des cellules témoins au jour 6 (D6, orange) et au jour 11 (D11, bleu), après fusion des deux jeux de données, illustrant les dynamiques de différenciation. Cette projection met en évidence les dynamiques de différenciation des cellules au cours du processus de mégacaryopoïèse. La réduction de dimension simplifie les données complexes à haute dimension (des milliers de gènes) en un espace bidimensionnel, tout en préservant les relations essentielles entre les cellules.
B – UMAP des cellules témoins après clustering (résolution = 1,2). Chaque couleur représente un cluster distinct.
C – Diagramme en points montrant les niveaux d’expression des gènes spécifiques des cellules hématopoïétiques dans chaque cluster (analyse supervisée). La couleur reflète l’intensité moyenne d’expression relative par cluster ; la taille du point indique le pourcentage de cellules exprimant le gène.
D – UMAP des cellules témoins annotées selon les types cellulaires hématopoïétiques, en superposant les clusters défi nis en B. Chaque type cellulaire est coloré distinctement.
E – UMAP des cellules témoins après le calcul et le scoring des signatures proposées. Chaque graphique représente le score associé à une signature d’un type cellulaire donné. Le dégradé de couleur indique le score relatif de la signature.
F – UMAP combiné des cellules issues de témoins et de patients porteurs de variants ETV6, à D6 et D11, coloré selon les types cellulaires. Les populations identifiées précédemment ont été réassignées aux cellules porteuses de variant ETV6 à l’aide des codes-barres HTO. Des altérations significatives apparaissent à partir du stade MEP.
G – Schéma récapitulatif des voies dérégulées dans les MK de patients ETV6. Les cellules porteuses de variant ETV6 présentent une augmentation de la traduction et de l’activité ribosomale (notamment via la phosphorylation de RPS6), ainsi qu’une altération des voies de réparation de l’ADN. Ces modifications sont associées à un défaut de maturation mégacaryocytaire et de production plaquettaire.
Par ailleurs, l’application du même pipeline analytique à des patients porteurs de variants FLI1 a révélé une dérégulation attendue des voies d’agrégation plaquettaire, ainsi qu’un déficit en taline 1, jusqu’alors non décrit, soulignant l’intérêt de cette approche dans la compréhension fine des pathologies plaquettaires (7). Bien que le scRNA-seq soit souvent réservé à l’étude de pathologies ayant un fort retentissement transcriptomique, notamment celles impliquant des FT, son potentiel dépasse largement ce cadre. L’exploration des PPC associées à des anomalies dans des gènes du cytosquelette ou de la signalisation est tout aussi prometteuse, car ces protéines peuvent indirectement moduler l’expression génique, agir en tant que cofacteurs ou réguler l’activité des FT.
Un défi majeur reste l’interprétation biologique et la validation fonctionnelle des résultats issus du scRNA-seq. Si l’identification de gènes ou de voies dérégulées constitue une première étape, leur impact fonctionnel n’est pas toujours évident à définir. Les validations par qRT-PCR, immunoblot ou par des analyses fonctionnelles sur MK ou plaquettes sont indispensables, mais elles tendent à privilégier les gènes fortement exprimés au détriment de gènes faiblement exprimés mais essentiels. Des outils comme SCENIC permettent d’identifier les réseaux de régulation transcriptionnelle propres à certaines sous-populations de MK, en mettant en évidence les FT impliqués. À terme, ces analyses gagneraient à être complétées par des approches multi-omiques combinant transcriptomique, épigénomique et protéomique. Par exemple, le scATAC-seq, en reliant expression génique et accessibilité chromatinienne, pourrait déterminer si certains variants affectent l’ouverture de régions régulatrices clés pendant la différenciation mégacaryocytaire. Cette technique permet de cibler plus précisément les perturbations de l’activation génique. Enfin, l’approche protéomique apporterait une dimension fonctionnelle complémentaire, en identifiant les altérations des voies de signalisation ou des interactions protéine-protéine, contribuant ainsi à la découverte de nouvelles cibles diagnostiques ou thérapeutiques pour les PPC.
• La revue décrit une méthodologie pratique du scRNA-seq basée sur la technologie 10X Genomics, associée à des outils bio-informatiques, pour étudier la différenciation mégacaryocytaire à partir de cellules CD34+ cultivées in vitro.
• L’analyse transcriptomique des cultures de cellules CD34+ a permis d’identifier et de caractériser les différentes populations cellulaires impliquées dans la mégacaryopoïèse in vitro.
• Appliquée à la thrombopénie associée à des variants du gène ETV6, cette approche a révélé un blocage de la différenciation des mégacaryocytes, une augmentation de la voie de la traduction et une diminution des voies de réparation de l’ADN.
• L’intégration des données transcriptomiques à d’autres approches multiomiques pourrait affiner la compréhension des mécanismes physiopathologiques des pathologies plaquettaires constitutionnelles.
• Une collaboration étroite et continue entre bio-informaticiens et biologistes est essentielle pour garantir la fiabilité et la qualité des résultats issus de cette technologie.