RÉSUMÉ
L’acquisition de la masse osseuse au cours de l’enfance et de l’adolescence est un déterminant majeur du risque ultérieur d’ostéoporose et de fractures. Chez les enfants atteints d’hémophilie, plusieurs mécanismes peuvent compromettre cette acquisition et conduire à une diminution de la densité minérale osseuse (DMO) et à une fragilité osseuse accrue. Des données expérimentales et cliniques suggèrent que le déficit en facteur VIII (FVIII) ou en facteur IX (FIX) peut altérer directement le remodelage osseux, indépendamment des hémarthroses. Ces effets impliquent notamment une dysrégulation des voies RANK/RANKL/OPG et Wnt/ß-caténine, favorisant la résorption osseuse et réduisant la formation osseuse. Par ailleurs, la diminution de la génération de thrombine apparaît comme un mécanisme central reliant le déficit en facteurs de coagulation à l’altération du remodelage osseux, en raison de ses effets directs sur les ostéoblastes et les ostéoclastes. À ces mécanismes s’ajoutent des facteurs indirects, notamment les hémarthroses, qui induisent des altérations locales et inflammatoires, ainsi que la réduction des contraintes mécaniques liée à l’arthropathie hémophilique, contribuant à une diminution de la formation osseuse. Les facteurs nutritionnels, en particulier un statut insuffisant en vitamine D, peuvent également aggraver ces anomalies au cours de la croissance. Ainsi, la fragilité osseuse dans l’hémophilie résulte d’interactions complexes entre mécanismes biologiques directs et facteurs fonctionnels et environnementaux. Une meilleure compréhension de ces processus est essentielle pour optimiser les stratégies de prévention et de prise en charge de la santé osseuse chez les enfants atteints d’hémophilie.MOTS CLÉS
facteur VIII, fragilité osseuse, hémophilie, thrombine
ABSTRACT
Bone mass acquisition during childhood and adolescence is a major determinant of future osteoporosis and fracture risk. In children with hemophilia, several mechanisms may impair this process, leading to reduced bone mineral density (BMD) and increased bone fragility. Experimental and clinical data suggest that factor VIII (FVIII) or factor IX (FIX) deficiency may directly alter bone remodeling, independently of hemarthrosis. These effects involve dysregulation of key pathways, including the RANK/RANKL/OPG system and the Wnt/ß-catenin pathway, promoting bone resorption and reducing bone formation. In addition, decreased thrombin generation appears to be a central mechanism linking coagulation factor deficiency to impaired bone remodeling, through its direct effects on osteoblasts and osteoclasts. Indirect factors also contribute, including hemarthrosis, which induces local inflammatory changes, and reduced mechanical loading related to hemophilic arthropathy, both contributing to decreased bone formation. Nutritional factors, particularly insufficient vitamin D status, may further exacerbate these alterations during growth. Overall, bone fragility in hemophilia results from complex interactions between direct biological mechanisms and functional and environmental factors. A better understanding of these processes is essential to optimize strategies for the prevention and management of bone health in children with hemophilia.
KEYWORDS
bone fragility, factor VIII, hemophilia, thrombin
INTRODUCTION
La masse osseuse se construit durant l’enfance et l’adolescence, avec un pic atteint au début de l’âge adulte. Ce capital osseux, acquis à 90 % avant 18 ans, conditionne le risque ultérieur d’ostéoporose et de fractures.
Dans ce contexte, toute altération des processus de remodelage osseux ou de contrainte mécanique au cours de la croissance peuvent compromettre l’acquisition du capital osseux. Chez les enfants atteints d’hémophilie, plusieurs mécanismes sont susceptibles de contribuer à cette altération, incluant des effets directs liés au déficit en facteurs de coagulation, une diminution de la génération de thrombine, ainsi que les conséquences des saignements articulaires sur l’activité physique et la mobilité.
Par ailleurs, avec l’émergence de nouvelles thérapies, un intérêt croissant s’est porté sur les rôles extra-hémostatiques des facteurs de coagulation, en particulier leur implication dans la biologie osseuse.
Cette revue narrative propose une synthèse des mécanismes impliqués dans la diminution de la DMO et la fragilité osseuse chez l’enfant atteint d’hémophilie.
MÉCANISMES PHYSIOPATHOLOGIQUES DE LA FRAGILITÉ OSSEUSE DANS L’HÉMOPHILIE
Les rôles des facteurs de la coagulation dans l’homéostasie osseuse sont résumés et illustrés dans la figure 1.
Altération directe du remodelage osseux liée au déficit en FVIII/FIX
Plusieurs études expérimentales menées sur des modèles murins suggèrent que le FVIII pourrait contribuer à l’homéostasie osseuse, avec des effets mesurables sur la résorption osseuse et la DMO. Le déficit en FVIII pourrait affecter directement la masse osseuse indépendamment des hémarthroses et de l’activité physique (1).
À l’aide de souris invalidées pour le gène F8 (FVIII−/−), les auteurs ont comparé environ 20 mâles FVIII−/− à
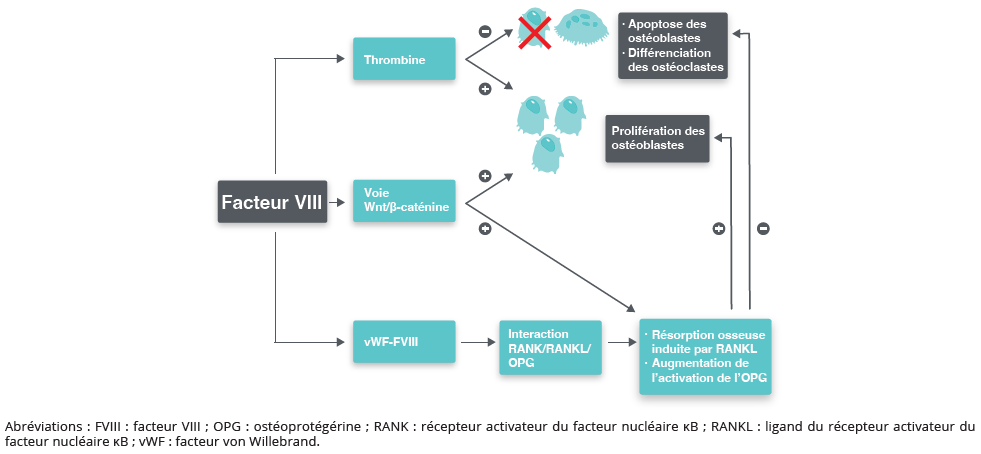
Figure 1 : Rôles des facteurs de la coagulation dans l’homéostasie osseuse.
Figure 1: Roles of coagulation factors in bone homeostasis.
des témoins de type sauvage à maturité squelettique (18-20 semaines). La DMO a été évaluée par DXA et la géométrie fémorale par microtomodensitométrie (µCT). Les souris FVIII−/− présentaient une DMO fémorale plus faible et une diminution de l’épaisseur corticale moyenne (p = 0,036 et p = 0,039, respectivement), associées à une réduction d’environ 10 % de la résistance à la fracture. Des diminutions de la rigidité fémorale, de la résistance intrinsèque de l’os et de son élasticité ont également été observées, soutenant une association entre le déficit en FVIII et une altération des propriétés mécaniques osseuses (1).
En 2013, Recht et al. ont approfondi l’étude des mécanismes potentiels à l’aide d’analyses biochimiques et histomorphométriques chez des souris FVIII−/− comparées à des témoins (2). Les marqueurs circulants de formation osseuse (ostéocalcine) et les médiateurs liés à l’ostéoclastogenèse (RANK et ostéoprotégérine) ne différaient pas entre les groupes, tandis que des cytokines impliquées dans la régulation du remodelage osseux, notamment l’IL-1α et l’IFN-β, étaient largement indétectables chez les souris FVIII−/− (p < 0,01 et p = 0,049). Les analyses histomorpho-métriques étaient compatibles avec une ostéopénie trabéculaire, caractérisée par une diminution de la surface osseuse et du nombre de travées, ainsi qu’une augmentation de leur séparation. Une augmentation du nombre d’ostéoclastes suggérait une résorption osseuse accrue.
Les données cliniques comparant les paramètres osseux entre les personnes atteintes d’hémophilie A (PwHA) et d’hémophilie B (PwHB) restent limitées, ces populations étant fréquemment analysées conjointement. Une augmentation de la résorption osseuse — reflétée par des niveaux plus élevés de télopéptides C-terminaux du collagène de type I (CTX-I) — a été rapportée en cas de déficit en FVIII mais non en cas de déficit en FIX, tandis que des concentrations plus élevées d’ostéoprotégérine (OPG) dans le plasma des sujets déficients en FIX pourraient contribuer aux différences observées de production de CTX-I entre les individus déficients en FVIII et en FIX (2).
Battafarano et al. ont étudié in vitro les effets directs de plusieurs facteurs de coagulation (FVIII, vWF, complexe FVIII/vWF, FIXa, FXa et thrombine) sur le comportement des ostéoclastes et des ostéoblastes, ainsi que l’impact de thérapies non substitutives, notamment l’emicizumab et les anticorps denecimig (Mim8), sur des sous-populations monocytaires servant de précurseurs ostéoclastiques (3).
Le FVIII, le vWF, le complexe FVIII/vWF, le FXa et la thrombine inhibaient l’ostéoclastogenèse, tandis que le FIXa n’exerçait aucun effet détectable sur la différenciation des ostéoclastes.
Ces résultats semblent diverger des données animales rapportées par Taves et al., dans lesquelles les souris FVIII−/− et FIX−/−, mais non les souris vWF−/−, développaient un phénotype ostéoporotique avec diminution de la DMO (4).
Dans l’ensemble, ces données suggèrent un effet direct du déficit en facteurs de coagulation sur le remodelage osseux, impliquant plusieurs voies moléculaires décrites ci-dessous.
Dysrégulation des voies du remodelage osseux
Inhibition de la voie RANK/RANKL/OPG
Le système RANK/RANKL/OPG joue un rôle central dans la régulation de la différenciation des ostéoclastes et de la résorption osseuse. Le ligand du récepteur activateur du facteur nucléaire κB (RANKL), exprimé par les ostéoblastes et les cellules stromales, se lie à son récepteur RANK sur les précurseurs ostéoclastiques, favorisant leur maturation et leur activation. L’OPG agit comme un récepteur leurre du RANKL, inhibant ainsi l’ostéo-clastogenèse.
Des données expérimentales suggèrent que le FVIII pourrait moduler cette voie. Il pourrait influencer l’équilibre entre RANKL et OPG, et son déficit entraîner une augmentation de l’activité du RANKL et une diminution des niveaux d’OPG, favorisant la résorption osseuse. Chez des souris ayant un déficit en FVIII, l’administration de FVIII réduit d’environ 25 % l’expression de RANKL lors de la différenciation des cellules souches mésenchymateuses médullaires en ostéoblastes, soutenant un effet direct du FVIII sur l’homéostasie osseuse via cette voie. Des études cliniques chez les patients atteints d’hémophilie ont également montré des taux circulants plus élevés de RANKL et plus faibles d’OPG que chez les témoins, suggérant une augmentation de l’activité ostéoclastique (5).
Inhibition de la voie Wnt/β-caténine
La voie de signalisation Wnt/β-caténine est un régulateur clé de la différenciation des ostéoblastes et de la formation osseuse. L’activation de cette voie favorise la prolifération et la fonction des ostéoblastes, tandis que son inhibition entraîne une diminution de la formation osseuse. La sclérostine, une glycoprotéine produite par les ostéocytes, inhibe la voie Wnt et régule négativement la formation osseuse. Des taux circulants élevés de sclérostine ont été rapportés chez les enfants atteints d’hémophilie, suggérant une inhibition de l’activité ostéoblastique et une contribution potentielle à la diminution de la DMO (6).
Déficit en thrombine : un mécanisme central
En 2019, Taves et al. ont comparé la structure osseuse de base et les réponses du remodelage après hémarthrose chez des souris FVIII−/−, FIX−/− et vWF−/− à l’aide de mesures issues de la DXA et de la µCT (4). Les souris FVIII−/− et FIX−/− présentaient des déficits comparables de DMO et de microarchitecture trabéculaire, qui s’accentuaient après hémarthrose. Des résultats concordants ont été rapportés par Larson et al. dans un modèle d’hémophilie B, montrant une DMO plus faible, une diminution des indices de masse osseuse corticale et trabéculaire, ainsi qu’une réduction de la résistance fémorale à la fracture chez les souris FIX−/− (7). Notamment, ce phénotype osseux anormal n’était pas observé chez les souris vWF−/−, suggérant que le complexe FVIII/vWF n’est pas indispensable en soi au maintien de l’intégrité osseuse après une lésion. Ces observations suggèrent plutôt que la préservation de la génération de thrombine chez les souris vWF−/− pourrait contribuer au maintien de l’homéostasie osseuse par rapport aux déficits en FVIII ou FIX (4).
Plusieurs études soulignent par ailleurs le rôle central de la thrombine dans l’homéostasie osseuse (8). Les ostéoblastes et les ostéoclastes expriment des récepteurs de la thrombine, notamment le récepteur activé par les protéases de type 1 (PAR1). In vitro, la thrombine favorise la différenciation des ostéoblastes et la formation osseuse, tout en inhibant le développement et l’activité de résorption des ostéoclastes. De façon concordante, Aronovitch et al. ont montré que des souris présentant une inactivation complète du FVIII ou de PAR1 développent des anomalies structurelles osseuses similaires, soutenant un lien mécanistique entre une altération de la génération de thrombine et une perturbation du remodelage osseux.
La diminution de la génération de thrombine apparaît donc comme un mécanisme central reliant le déficit en FVIII/FIX à l’altération du remodelage osseux.
Effets indirects liés aux hémarthroses
Des travaux ultérieurs ont étudié l’impact de la substitution en FVIII dans un contexte d’hémarthrose aiguë (9). Après ponction de l’articulation du genou chez des souris FVIII−/−, les animaux ont reçu du FVIII par voie intraveineuse ou un placebo immédiatement avant la lésion, puis à 4, 24, 48, 72 et 96 heures. Une perte osseuse tibiale proximale et une calcification des tissus mous étaient observées chez les souris traitées par placebo, tandis que l’administration de FVIII atténuait significativement à la fois la perte osseuse et la dégénérescence des tissus périarticulaires. Ces résultats soutiennent le rôle de l’hémarthrose aiguë dans la survenue précoce de la perte osseuse en l’absence de traitement substitutif en FVIII (9).
Ces données suggèrent que les saignements articulaires contribuent à la fragilité osseuse via des mécanismes inflammatoires et locaux, en plus des effets directs du déficit en facteurs de coagulation.
Diminution des contraintes mécaniques et immobilisation
La réduction des contraintes mécaniques et l’immobilisation prolongée constituent des facteurs bien reconnus de perte osseuse et d’ostéoporose. Chez les patients atteints d’hémophilie, plusieurs études ont montré une association inverse entre la DMO et la sévérité de l’arthropathie hémophilique, suggérant que la limitation de l’activité liée aux atteintes articulaires peut altérer la santé osseuse. Par ailleurs, l’augmentation des taux de sclérostine observée chez les enfants atteints d’hémophilie renforce l’hypothèse d’un rôle de la diminution des contraintes mécaniques dans l’altération de la formation osseuse (6).
Carence en calcium et vitamine D
La vitamine D joue un rôle essentiel dans l’absorption du calcium et la minéralisation osseuse. Elle est principalement synthétisée au niveau de la peau sous l’effet du rayonnement ultraviolet B, l’apport alimentaire ne représentant qu’une contribution moindre. Un statut adéquat en vitamine D au cours de la croissance constitue un déterminant important de la minéralisation du squelette et est associé à des valeurs plus favorables de DMO chez l’enfant. En conséquence, des concentrations sériques plus faibles de 25-hydroxyvitamine D [25(OH)D] ont été associées à un risque accru d’altération de la santé osseuse dans les populations pédiatriques (10).
POINTS CLÉS À RETENIR
• Le déficit en FVIII/FIX altère le remodelage osseux, en partie via une diminution de la génération de thrombine.
• La dysrégulation des voies RANK/RANKL/OPG et Wnt/ß-caténine pourrait favoriser la résorption osseuse chez les patients atteints d’hémophilie.
• Les hémarthroses, la réduction des contraintes mécaniques et les facteurs nutritionnels contribuent à la baisse de la DMO.