Actualités scientifiques
RÉSUMÉ
La Revue Francophone d’Hémostase et Thrombose vous propose, outre la réception trimestrielle de la revue, des publications online régulières sur la plateforme www.rfht.fr sous la forme : • d’une veille bibliographique avec les meilleurs articles publiés dans nos spécialités ; • d’actualités commentées offrant une vision critique d’une sélection d’articles internationaux et leur mise en perspective en pratique clinique. Vous trouverez ci-après une sélection de ces articles récents, décryptés, analysés et commentés pour vous par Wafa Amara, Alexandre Guy, Mouna Mahmoud et Agnès Ribes.En partenariat avec


La Revue Francophone d’Hémostase et Thrombose vous propose, outre la réception trimestrielle de la revue, des publications online régulières sur la plateforme www.rfht.fr sous la forme :
• d’une veille bibliographique avec les meilleurs articles publiés dans nos spécialités ;
• d’actualités commentées offrant une vision critique d’une sélection d’articles internationaux et leur mise en perspective en pratique clinique.
Vous trouverez ci-après une sélection de ces articles récents, décryptés, analysés et commentés pour vous par Wafa Amara, Alexandre Guy, Mouna Mahmoud et Agnès Ribes.
APIXABAN À DOSE RÉDUITE DANS LE TRAITEMENT PROLONGÉ DES MTEV ASSOCIÉES AU CANCER
D’après : Extended Reduced-Dose Apixaban for Cancer-Associated Venous Thromboembolism. Mahé I, Carrier M, Mayeur D, Chidiac J, Vicaut E, Falvo N, et al.
N Engl J Med 2025 ; 392 : 1363-73.
Actualité commentée réalisée par Wafa AMARA
Justificatifs et objectifs
Chez les patients atteints de cancer ayant présenté une maladie thromboembolique veineuse (MTEV), une anticoagulation par un anticoagulant oral direct (AOD) ou héparine de bas poids moléculaire (HBPM) est recommandée pour une durée minimale de 6 mois. Au-delà de cette période, la poursuite du traitement anticoagulant est recommandée tant que le cancer est actif ou que le traitement anticancéreux est en cours, tenant compte du risque thrombotique et hémorragique du patient. Cependant, la modalité du traitement anticoagulant prolongé (i.e. au-delà de 6 mois) reste incertaine. L’objectif de cette étude était d’évaluer si une dose réduite d’apixaban (2,5 mg x 2/j) est non inférieure à la dose standard (5 mg x 2/j) pour prévenir les récidives de MTEV chez des patients cancéreux après 6 mois de traitement anticoagulant initial.
Méthodes
API-CAT est une étude multicentrique, internationale, randomisée, en double aveugle, de non-infériorité, incluant 1 766 patients atteints de cancer actif (sein, prostate, colon ou rectum, poumons ou autres) et ayant reçu ≥ 6 mois d’anticoagulation pour une MTEV sans récidive (thrombose veineuse proximale du membre inférieur et/ou embolie pulmonaire symptomatique ou de découverte fortuite au moins segmentaire). Les participants ont été randomisés pour recevoir, pour une durée supplémentaire de 12 mois, de l’apixaban à dose réduite (2,5 mg x2/j) ou standard (5 mg x2/j).
Le critère principal d’efficacité était la récidive de MTEV. Le critère principal de sécurité était les hémorragies cliniquement significatives (majeures ou non majeures cliniquement pertinentes selon la définition de l’ISTH). Les principaux critères d’exclusion comprenaient une espérance de vie < 3 mois, une thrombopénie < 75 G/L et une insuffisance rénale sévère (DFG < 30 ml/min, Cockcroft-Gault).
Résultats
Tableau 1 : Principales caractéristiques du TCA et de l’anti-Xa pour le suivi du traitement par HNF.
Table 1: Main characteristics of TCA and anti-Xa for monitoring treatment with UFH.
Table 1: Main characteristics of TCA and anti-Xa for monitoring treatment with UFH.
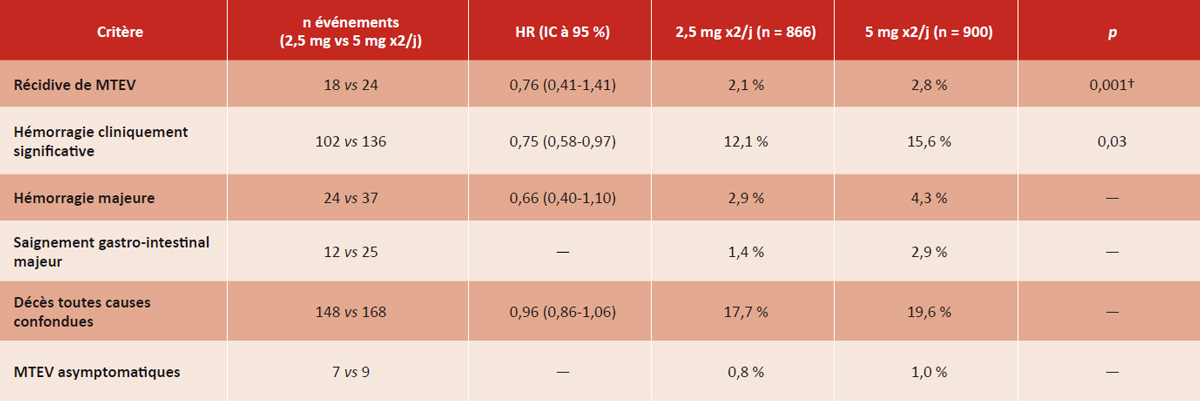
† p pour la non-infériorité (critère principal)
Avis d’expert
L’étude API-CAT répond à une question clinique fréquente et pertinente : quelle stratégie adopter après 6 mois d’anticoagulation chez les patients atteints de cancer ayant présenté une MTEV pour prévenir les récidives ? L’étude démontre que l’efficacité de l’apixaban à dose réduite est non inférieure à celle de la dose standard, avec une réduction significative du risque hémorragique, notamment majeur et gastro-intestinal.
La méthodologie est solide, avec une randomisation rigoureuse, une adjudication indépendante et une bonne représentativité de la population de patients cancéreux rencontrés en pratique clinique. Cependant, la majorité des patients inclus avaient un bon état général (92 % ECOG 0-1), des cancers stables ou contrôlés, et une faible proportion sous chimiothérapie, limitant l’extrapolation des résultats aux patients plus sévères ou instables.
En comparaison, l’étude EVE (1), bien qu’ouverte et non comparative, avait montré des résultats similaires, une faible incidence de récidive et de saignement sous apixaban 2,5 mg x 2/j chez des patients atteints de cancer stable. La présente étude randomisée et contrôlée apporte un niveau de preuve supérieur.
Par ailleurs, la durée de suivi reste courte (12 mois), ne permettant pas de trancher sur la stratégie à plus long terme. Le taux de récidive très faible dans les deux bras (environ 2 %) interroge sur la pertinence de poursuivre une anticoagulation prolongée chez certains patients à faible risque thrombotique. Des études complémentaires restent nécessaires pour évaluer le rapport bénéfice/risque de la poursuite d’un traitement anticoagulant chez des sous-groupes spécifiques de patients cancéreux au-delà des
6 premiers mois de traitement après une MTEV.
RÉFÉRENCE
1. McBane RD, Loprinzi CL, Zemla T, Tafur A, Sanfilippo K, Liu JJ, et al. Extending venous thromboembolism secondary prevention with apixaban in cancer patients. The EVE trial. J Thromb Haemost 2024 ; 22 : 1704-14.
ÉTUDE À L’ÉCHELLE DE POPULATIONS D’ANOMALIES DE LA PROTÉINE S ET DU RISQUE DE THROMBOSE
D’après : Population-Scale Studies of Protein S Abnormalities and Thrombosis. Chaudhry SA, Haj AK, Ryu J, Jurgens SJ, Rodriguez Espada A, Wang X, et al. JAMA 2025 ; 333 : 1423-32.
Actualité commentée réalisée par Alexandre GUY
Justificatifs et objectifs
La protéine S (PS) est une protéine plasmatique, vitamine K-dépendante, impliquée dans la régulation négative de la coagulation. Il existe trois formes de déficit héréditaire en protéine S : quantitatif (type I), qualitatif (type II), et un déficit en PS libre, mais pas en PS totale (type III). Le sur-risque de maladie thrombo-embolique veineuse (MTEV) induit par la présence d’un déficit en PS est controversé du fait de biais méthodologiques fréquemment retrouvés dans les études. Les objectifs de cette étude étaient de (i) caractériser les variants hétérozygotes « perte de fonction » au niveau du gène PROS1, (ii) d’évaluer l’association entre la présence de variants et le risque thrombotique, (iii) d’analyser l’association entre les taux circulants de PS et le risque de MTEV, (iv) d’analyser la prévalence du déficit en PS héréditaire au sein d’une large population.
Méthodes
Les auteurs ont analysé les données d’exome et/ou de génome entier issues de la base UK Biobank (n = 426 436) et de la base américaine du NIH (n = 204 006). Des données de protéomiques étaient disponibles pour 54 219 participants de la base UK Biobank. Des méthodes in silico ont été utilisées pour prédire le pouvoir pathogène des variants mis en évidence et l’établissement d’un score fonctionnel (functional impact score, FIS) mis au point. Ce score, compris entre 0 et 1, indiquait une pathogénicité plus importante à mesure que sa valeur se rapprochait de 1.
Les auteurs ont également analysé l’association entre la présence de variants rares (fréquence allélique < 0,1 %) et la survenue de différentes pathologies thrombotiques. Les variants rares avec un score FIS > 0,7 étaient considérés comme fonctionnellement délétères. Pour 44 431 participants, une étude de la concentration en PS libre a été réalisée par la méthode Olink Explore. Enfin, les auteurs ont déterminé la prévalence du déficit héréditaire en PS au sein des deux bases anglaise et américaine.
Résultats
La prévalence de patients avec des variants qui possèdent un score FIS > 0,7 ou égale à 1 était faible, estimée à respectivement 0,22 % et 0,0091 % des cas. Toutefois, la présence d’un variant hétérozygote de PROS1 dont le FIS était supérieur à 0,7 était associée à une augmentation significative du risque de MTEV (OR, 1,977 ; IC 95 %, 1,552-2,483) après ajustement sur l’âge, le sexe et la descendance. Cette association était encore plus marquée en cas de variant dont le FIS était égal à 1 (OR, 14,01 ; IC 95 %, 6,98-27,14). En revanche, aucune association n’a été observée entre ces variants et la survenue de thrombose artérielle. La présence d’un variant hétérozygote était également associée à une incidence cumulative significativement plus élevée de MTEV, tant pour les variants avec un FIS > 0,7 que pour ceux avec un FIS = 1,comparativement aux sujets ne présentant pas de mutation du gène PROS1. Fait intéressant, les variants avec un FIS > 0,7 étaient associés à une réduction très modérée et non significative des taux de protéine S (PS) en analyse protéomique (médiane : 97 %). En revanche, les trois patients porteurs d’un variant avec un FIS = 1 inclus dans cette analyse présentaient une diminution significative du taux de PS, avoisinant 50 %.
Concernant les données de protéomique, les auteurs ont mis en évidence une association entre une diminution du taux de PS et le risque de MTEV (pour des taux inférieurs à 60 % : OR, 2,50 ; IC 95 %, 1,80-3,46), mais aussi avec la survenue d’une artériopathie oblitérante des membres inférieurs. A noter que seuls 10,1 % des sujets avec un taux de PS < 60 % avaient des variants de PROS1 identifiés, suggérant la présence de causes acquises dans une majorité des cas de diminution de PS. Les auteurs ont ensuite validé ces résultats dans la cohorte américaine (n = 204 006) retrouvant des associations similaires entre les variants avec un score FIS > 0,7 ou FIS = 1 et le risque accru de MTEV, ainsi qu’une incidence plus élevée de MTEV au cours du suivi chez les porteurs de variants pathogènes.
Avis d’expert
Cette étude est la plus large conduite sur la présence de déficits en PS via l’étude de deux cohortes de taille très importante. Les auteurs retrouvent une prévalence faible de déficits constitutionnels en PS dans les 2 cohortes étudiées. Toutefois, la présence de variants pathogènes de PROS1 est associée à un risque significatif de MTEV plus élevé que celui décrit précédemment. Ces données suggèrent que des diminutions du taux antigénique de PS sont principalement médiées par des causes acquises et que, même en l’absence de variant identifié, la présence d’une diminution du taux de PS est associée à un risque augmenté de MTEV.
Une des forces de cette étude réside dans l’approche intégrée combinant des analyses in silico et la validation par des données de protéomique. Ces données suggèrent donc qu’il est pertinent en pratique clinique d’effectuer un dosage de la PS couplé à une étude du gène de la PS. En effet, parmi les patients avec diminution de la PS, ceux porteurs de variants hétérozygotes de PROS1 avec un score FIS élevé présentent un risque accru de MTEV. Ces données suggèrent enfin un intérêt limité de rechercher un déficit héréditaire en PS dans un contexte de thrombose artérielle.
ESTIMATION MONDIALE DES ÉPISODES HÉMORRAGIQUES TRAITABLES PAR LA DESMOPRESSINE DANS LA MALADIE DE VON WILLEBRAND ET L’HÉMOPHILIE A
D’après : Global estimation of the bleeding episodes treatable with desmopressin in von Willebrand disease and hemophilia A. Gringeri A, Mannucci PM, Gringeri M, Castaman G, Peyvandi F,
et al. Haematologica 2025 ; 110 : 1710-22.
Actualité commentée réalisée par Mouna MAHMOUD
Justificatifs et objectifs
La maladie de von Willebrand de type 1 (VWD1) et l’hémophilie A non sévère sont les troubles héréditaires de la coagulation les plus communs. La desmopressine est une thérapie efficace, abordable et sûre, bien que son utilisation soit encore restreinte. Les auteurs ont réalisé une étude statistique afin d’évaluer le nombre de patients qui pourraient profiter de ce traitement ainsi que le nombre d’épisodes hémorragiques qui pourraient être soignés chaque année. Dans le contexte mondial actuel où l’accès aux traitements de substitution est inégal de par les coûts engagés, cette question est d’une importance cruciale.
Méthodes
Les auteurs ont utilisé MEDLINE et d’autres bases de données (Cochrane, Google Scholar, registres nationaux) avec comme mots-clés les termes MESH, afin d’estimer la prévalence des formes non sévères de VWD et d’hémophilie A, l’incidence des saignements, et les taux de réponse à la desmopressine. Les données ont été croisées avec les estimations démographiques des Nations Unies et les classifications économiques de la Banque Mondiale pour modéliser l’impact potentiel du traitement au niveau mondial.
Résultats
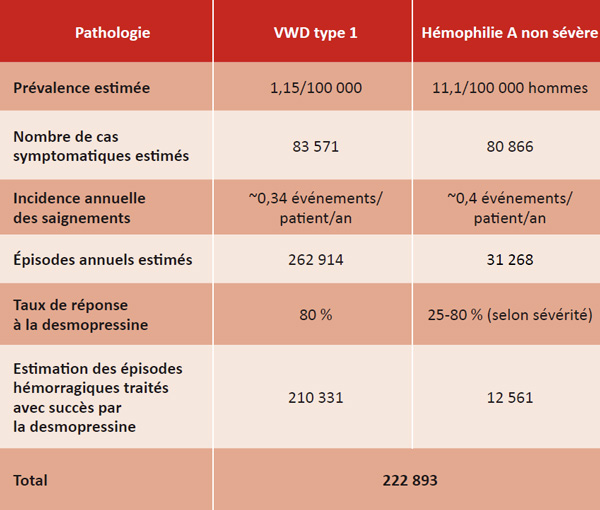
Avis d’expert
Cet article propose une estimation du nombre d’épisodes hémorragiques qui pourraient être traités avec la desmopressine chez les personnes atteintes de VWD1 ou d’hémophilie A non sévère. Ce sont deux troubles de la coagulation fréquents, et l’étude montre que la desmopressine, pourtant efficace et peu coûteuse, est encore très peu utilisée, surtout dans les pays à faibles revenus.
Les auteurs ont utilisé différentes sources, comme des articles scientifiques, des registres et des bases de données, pour tenter d’évaluer combien de patients pourraient bénéficier de ce traitement dans le monde. Afin que les résultats restent fiables, ils ont choisi de prendre les estimations les plus conservatrices en comptant les cas les plus probables. L’article rappelle aussi les différences importantes dans l’accès aux soins selon les pays, ce qui change beaucoup la situation réelle.
Même si l’étude présente plusieurs points positifs, elle a aussi quelques limites. Menée de façon non systématique, la sélection des données peut prêter à controverse. Certains usages possibles de la desmopressine n’ont pas été pris en compte, comme la prévention des saignements avant une opération ou l’utilisation d’aide au diagnostic. De plus, les femmes porteuses de l’hémophilie A, qui peuvent pourtant présenter des symptômes, ne sont pas incluses dans l’analyse, ce qui est regrettable.
Néanmoins, ce travail aide à mieux comprendre le rôle que pourrait jouer la desmopressine dans le traitement des troubles de la coagulation, en particulier dans des zones géographiques où les autres traitements sont rares ou trop coûteux.
UN VARIANT GAIN DE FONCTION DE RSG18 CANDIDAT POUR UN SYNDROME HÉMORRAGIQUE FAMILIAL MODÉRÉ
D’après : A gain-of-function variant in RGS18 candidate for a familial mild bleeding syndrome. Vayne C, Roux M, Gruel Y, Poggi M, Pouplard C, Peiretti F, et al. J Thromb Haemost 2025 ; 23 : 314-20.
Actualité commentée réalisée par Agnès RIBES
Justificatifs et objectifs
Les pathologies plaquettaires héréditaires affectant la production ou les fonctions plaquettaires demeurent rares et souvent insuffisamment caractérisées, en particulier lorsque le phénotype est modéré, sans anomalies morphologiques ni thrombopénie associée. En dépit des avancées technologiques récentes des explorations moléculaires, notamment en matière de séquençage à haut débit et d’analyse bio-informatique, un nombre encore trop important de patients reste sans diagnostic génétique établi.
Cette étude a pour objectif de caractériser un syndrome hémorragique familial modéré selon une approche intégrée, moléculaire, morphologique et fonctionnelle, et de proposer un mécanisme physiopathologique plausible.
Méthodes
Des patients suivis aux CHU de Bordeaux et de Tours ont été inclus après consentement éclairé, et approbation éthique (étude EVGPP). Un séquençage complet de l’exome (WES) a été réalisé chez trois individus atteints et un sujet sain apparenté. L’analyse a ciblé les variants codants rares potentiellement pathogènes exprimés dans la lignée plaquettaire. Le variant RGS18 a été recherché chez neuf membres de la famille par séquençage Sanger. Les fonctions plaquettaires ont été explorées par des tests standardisés. Des analyses statistiques et un alignement inter-espèce de la protéine RGS18 ont complété l’étude.
Résultats
Le séquençage de l’exome et les études de co-ségrégation ont identifié un variant hétérozygote RGS18 c.643C>T (p.Arg215*) chez 6 membres atteints d’une même famille. RGS18, fortement exprimé dans les plaquettes, agit comme un activateur de l’activité GTPasique de la sous-unité alpha des protéines G hétérotrimériques et donc comme un régulateur négatif des voies de signalisation des récepteurs couplés aux protéines G (RCPG). La mutation introduit un codon-stop prématuré, supprimant des sites de phosphorylation critiques (S216, S218) nécessaires à son interaction avec la protéine 14-3-3 qui neutralise son activité (Figures 1 et 2).
Les patients analysés présentaient une diathèse hémorragique sans thrombopénie, ni anomalie morphologique ou d’expression des principales glycoprotéines. L’agrégation plaquettaire était diminuée en réponse aux agonistes des RCPG (ADP, thrombine, Thromboxane A2, acide arachidonique, épinéphrine) en lien avec un défaut de sécrétion des granules denses. La réponse au collagène était conservée.
Cette forme constitutivement active gain-de-fonction représente le premier cas de thrombopathie héréditaire liée à RGS18, illustrant son rôle central dans l’homéostasie plaquettaire.
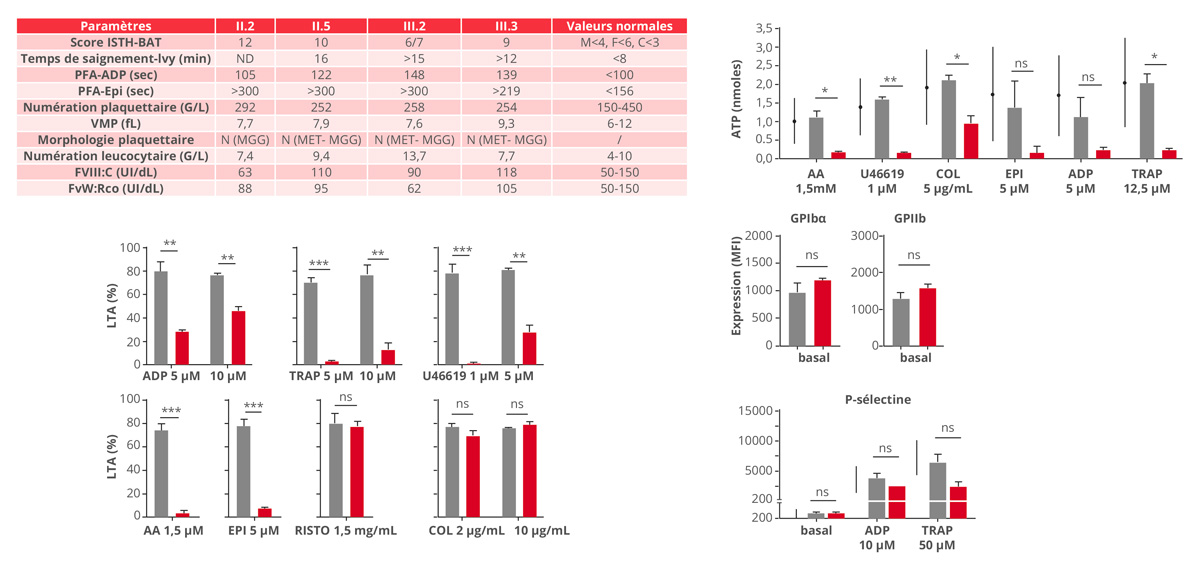
Abréviations : LTA : agrégométrie par transmission lumineuse ; ISTH-BAT : ISTH-bleeding assessment tool ; PFA : platelet function analyzer ; VMP : volume moyen plaquettaire ; FVIII : facteur VIII ; FvW : facteur Willebrand ; M : homme ; F : femme ; C : enfant ; GP : glycoprotéine.
Figure 1 : A. Tableau récapitulant les caractéristiques clinico-biologiques des membres de la famille portant RGS18 c.643C>T (p.Arg215*) ; B. Tests d’agrégation plaquettaire en réponse à l’ADP, la thrombine (TRAP), le thromboxane A2 (U46619), l’acide arachidonique (AA), l’épinéphrine (Epi), la ristocétine (RISTO) et le collagène (COL) ; C. Mesure de l’ATP libéré par les granules denses en réponse aux mêmes agonistes par lumino-agrégométrie ; D. Niveau d’expression basal des principales glycoprotéines plaquettaires et de l’expression de la P-sélectine (granules alpha) en réponse à l’ADP et un peptide analogue de la thrombine (TRAP).
Avis d’expert
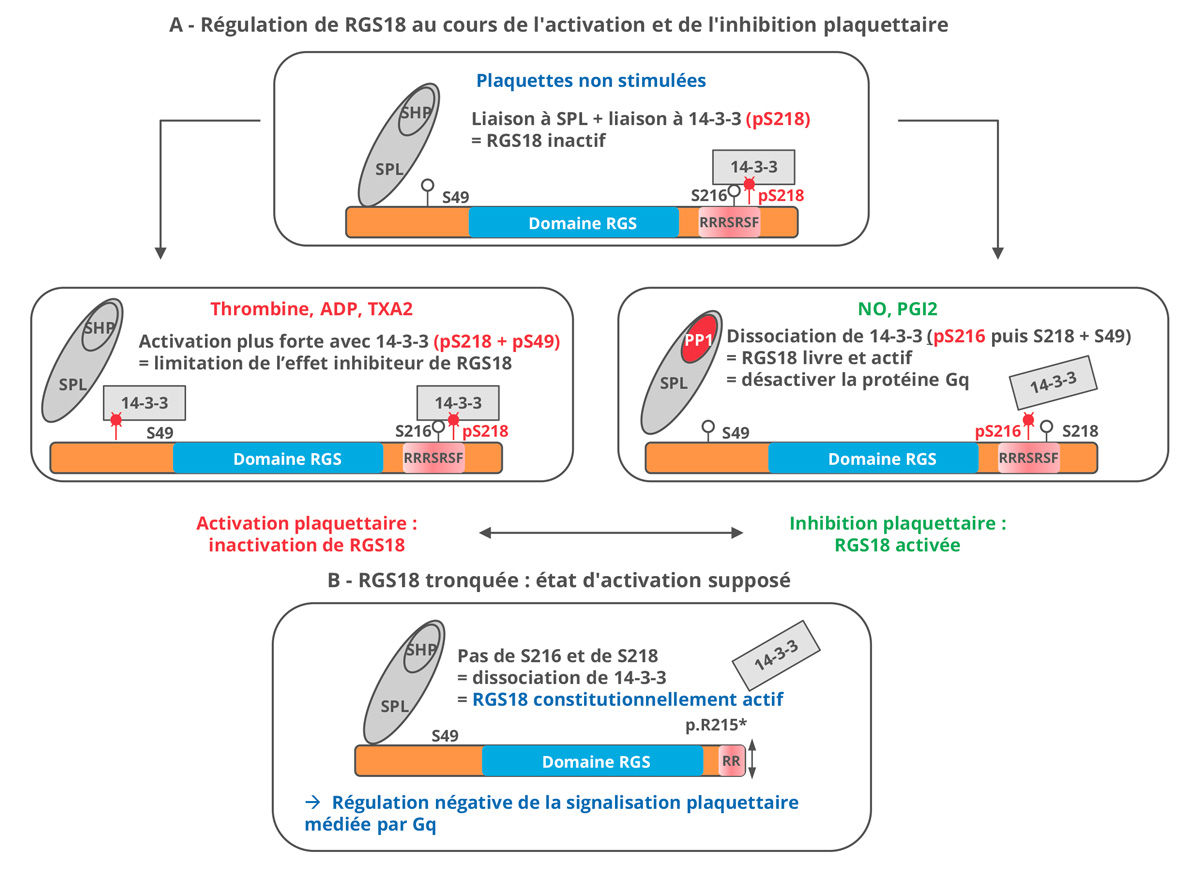
Figure 2 : Représentation schématique de la régulation de la protéine RGS18 native (A) et tronquée (B).
Au repos, RGS18 forme un complexe hétérotrimérique avec la spinophiline (SPL) et la tyrosine phosphatase SHP. Cette dernière se fixe à SPL par la phosphorylation du résidu Y398. RGS18 interagit également avec la protéine 14-3-3 via la phosphorylation du résidu S218.
Lors de l’activation plaquettaire par la thrombine, l’ADP ou le Thromboxane A2 (TxA2), la phosphorylation du résidu S49 vient renforcer l’interaction de la protéine 14-3-3 avec RGS18 et son inhibition. En parallèle, ces agonistes induisent la dissociation du complexe SPL-SHP/RGS18 par activation de SHP. RGS18 ne peut plus favoriser l’hydrolyse du GTP en GDP par la sous-unité Gαi/q, entraînant une augmentation de la signalisation en aval de ces RCPG. La modulation de la durée et l’intensité de ce signal sont méconnues.
Au repos, RGS18 forme un complexe hétérotrimérique avec la spinophiline (SPL) et la tyrosine phosphatase SHP. Cette dernière se fixe à SPL par la phosphorylation du résidu Y398. RGS18 interagit également avec la protéine 14-3-3 via la phosphorylation du résidu S218.
Lors de l’activation plaquettaire par la thrombine, l’ADP ou le Thromboxane A2 (TxA2), la phosphorylation du résidu S49 vient renforcer l’interaction de la protéine 14-3-3 avec RGS18 et son inhibition. En parallèle, ces agonistes induisent la dissociation du complexe SPL-SHP/RGS18 par activation de SHP. RGS18 ne peut plus favoriser l’hydrolyse du GTP en GDP par la sous-unité Gαi/q, entraînant une augmentation de la signalisation en aval de ces RCPG. La modulation de la durée et l’intensité de ce signal sont méconnues.
En présence d’un signal inhibiteur de l’activation plaquettaire, via la prostacycline (PGi2) ou l’oxyde nitrique (NO), les voies PKA et PKG sont induites. Le résidu S216 est phosphorylé alors que les résidus S49 et S218 sont déphosphorylés par la phosphatase 1 (PP1) liée à SPL. RGS18 se retrouve dissociée de la protéine 14-3-3 et en capacité de favoriser l’hydrolyse du GTP en GDP par la sous-unité Gαi/q et reformer le complexe hétérotrimérique de protéine Gi/q inactif.
Lorsque RGS18 est tronquée au niveau du motif RRRSRSF, juste avant les résidus S216 et S218, les interactions activatrice et inhibitrice avec la protéine 14-3-3 sont impossibles. La perte du résidu S218 empêche toute interaction avec la protéine 14-3-3 et bloque RGS18 en position constitutivement active augmentantl’hydrolyse du GTP.
Lorsque RGS18 est tronquée au niveau du motif RRRSRSF, juste avant les résidus S216 et S218, les interactions activatrice et inhibitrice avec la protéine 14-3-3 sont impossibles. La perte du résidu S218 empêche toute interaction avec la protéine 14-3-3 et bloque RGS18 en position constitutivement active augmentantl’hydrolyse du GTP.
Cette étude présente les éléments diagnostiques d’un nouveau variant hétérozygote dans le gène RGS18 (c.643C>T;p.Arg215*) identifié par séquençage de l’exome au sein d’une même famille. L’analyse combine les données cliniques (score hémorragique) aux données biologiques (numération plaquettaire, temps d’occlusion plaquettaire, exploration du facteur Willebrand, agrégation plaquettaire, lumino-agrégométrie, cytométrie en flux). La combinaison des analyses génotype/phénotype intra-familiales permet d’affirmer le rôle pathogène de cette nouvelle mutation hétérozygote. Enfin, cet article récapitule les éléments de régulation de RGS18 native et de sa forme tronquée dans un schéma intégratif permettant de mieux comprendre cette nouvelle thrombopathie constitutionnelle sans thrombopénie associée. Le spectre des gènes impliqués dans les troubles héréditaires plaquettaires s’élargit et inclut désormais les régulateurs de la signalisation intra-plaquettaire dans les panels diagnostiques.
Les points forts de ce travail, outre sa rigueur, sont la pluridisciplinarité. En effet, les données cliniques sont associées aux explorations de biologie moléculaire et aux analyses fonctionnelles. De plus, une interprétation des conséquences fonctionnelles du variant identifié est reprise dans le schéma illustrant le mécanisme physiopathologique présumé comme montré dans la figure 2 de l’article et repris ici. Les limites de cette étude résident dans les conséquences fonctionnelles exactes de la protéine RGS18 tronquée qui auraient pu être davantage explorées, notamment par des modèles cellulaires transfectés voire des modèles murins. Il aurait été intéressant de quantifier la protéine RGS18 tronquée afin de s’assurer de sa stabilité, et éventuellement de réaliser des co-immunoprécipitations de RGS18 avec la protéine 14-3-3 pour démontrer la perte de cette interaction par la forme tronquée de RGS18. La mobilisation du calcium intra-plaquettaire aurait pu être mesurée après stimulation par la thrombine, le U46619 ou l’ADP pour confirmer l’excès d’inhibition par RGS18 tronquée. De même, il aurait été intéressant de doser l’AMPc après stimulation par la PGi2 pour évaluer l’effet de RGS18 tronquée sur Gαs. Les partenaires de signalisation intracellulaire de RGS18 et les voies secondairement activées auraient également pu être étudiées (1). Les modèles cellulaires de transfection auraient pu apporter des informations quant au ratio entre RGS18 normale et tronquée responsable du phénotype. Selon les ressources, un modèle murin reproduisant la mutation aurait permis de confirmer sa pathogénicité et de décrire son impact de façon plus exhaustive ex vivo et in vivo (2).
En conclusion, l’apport de ce travail est significatif puisqu’il met en lumière une forme inédite de thrombopathie familiale, avec un niveau de preuve suffisant, et décrypte le mécanisme moléculaire probable.
RÉFÉRENCES
1. Naguy Z and Smolebski A. Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses. Res Pract Thromb Haemost 2018 ; 2 : 558-71.
2. DeHelian D, Gupta S, Wu J, Thorsheim C, Estevez B, Cooper M, et al. RGS10 and RGS18 differentially limit platelet activation, promote platelet production, and prolong platelet survival. Blood 2020 ; 136 : 1773-82.