RÉSUMÉ
Ce cas clinique décrit un nourrisson atteint d’un syndrome de Binder, associé à une chondrodysplasie ponctuée et à un déficit combiné en facteurs de coagulation vitamine K-dépendants, conduisant à la survenue d’une hémorragie intracrânienne massive nécessitant un geste d’évacuation neurochirurgical. Ce tableau résulte d’une mutation homozygote du gène GGCX,MOTS CLÉS
déficits combinés en facteurs vitamine K-dépendants, gamma-carboxylation enzymatique, hémorragie intracrânienne., vitamine K
ABSTRACT
This case report describes an infant with Binder syndrome, associated with chondrodysplasia punctata and a combined deficiency in vitamin K-dependent clotting factors, leading to a massive intracranial hemorrhage requiring neurosurgical evacuation. The diagnosis of a homozygous mutation in the GGCX gene explains the clinical phenotype and the necessity for a regular oral vitamin K supplementation. The clinical outcome for this young patient is currently favorable. Differential diagnoses to consider in similar clinical presentations include vitamin K antagonist embryopathy and VKORC1 gene mutations. Finally, this genetic diagnosis enables genetic counseling and appropriate management for future pregnancies.
KEYWORDS
combined vitamin K-dependent clotting factors deficiency, enzymatic gammacarboxylation, intracranial hemorrhage., vitamin K
Introduction
Les syndromes rares combinant anomalies du développement crânio-facial, atteintes squelettiques et troubles de l’hémostase peuvent représenter un défi diagnostique en période néonatale. Le syndrome de Binder, ou dysostose maxillo-nasale, est une malformation congénitale caractérisée par une hypoplasie du tiers médian de la face. Lorsqu’il est associé à d’autres anomalies osseuses et à un trouble de l’hémostase, il peut révéler une pathologie génétique sous-jacente. Nous rapportons ici le cas d’un nourrisson porteur d’une mutation homozygote du gène GGCX, présentant ce tableau clinique complexe. Ce cas illustre les implications diagnostiques, thérapeutiques et génétiques d’un tel déficit, ainsi que la nécessité d’un suivi rapproché dès la période périnatale.
Cas clinique
Nous rapportons ici l’histoire d’un petit garçon, premier enfant d’un couple apparenté. Cette grossesse est marquée par la découverte anténatale d’une malformation faciale évocatrice d’un syndrome de Binder. Il s’agit d’une malformation congénitale responsable d’une anomalie de développement du tiers médian du massif facial, associant une hypoplasie des maxillaires et des os du nez.
Une amniocentèse est proposée mais non souhaitée par les parents. Un scanner anténatal est réalisé. Il confirme le syndrome de Binder et met en évidence une calcification osseuse fémorale supérieure gauche, sans autre anomalie associée. La grossesse se poursuit mais évolue vers une menace d’accouchement prématuré (MAP), avec rupture prématurée des membranes à 33 semaines d’aménorrhée. Cette MAP échappe à la tocolyse et l’accouchement a lieu à 33 SA + 3 jours. Ce bébé présente une détresse respiratoire néonatale qui va nécessiter une intubation pendant 5 jours suivie d’une ventilation non invasive pendant 5 jours supplémentaires. L’évolution clinique est ensuite favorable et il sort d’hospitalisation pour le domicile.
Dans le cadre de ce syndrome de Binder, il bénéficie d’une radiographie de squelette qui montre la présence de calcifications importantes au niveau de la trachée, du tarse, des vertèbres, du fémur. Une IRM cervico-occipitale est demandée mais non réalisée.
Dans le cadre du suivi de néonatologie, il va bénéficier d’une IRM cérébrale à l’âge de 2 mois normale et de potentiels évoqués auditifs normaux.
À l’âge de 4 mois, il va présenter un épisode fébrile avec gêne respiratoire et une rhinopharyngite modérée, associée à une diminution de l’appétit, à une perte de poids, évoluant sur une semaine. La maman consulte aux urgences à trois reprises pendant ces quelques jours.
Il présente ensuite une aggravation de son état associant une hypotonie et une somnolence qui le conduisent de nouveau aux urgences. Le tableau clinique est évocateur de méningite, la ponction lombaire réalisée retrouve un liquide céphalorachidien hémorragique, le scanner cérébral fait alors le diagnostic d’hématomes sous-duraux étendus, avec une atteinte fronto-pariéto-occipitale gauche, un hématome sous-dural frontal droit avec déviation de la ligne médiane et engagement temporal droit. Cet enfant est ensuite transféré en neurochirurgie pour évacuation de l’hématome. L’examen ophtalmologique, notamment le fond d’œil, est normal. L’imagerie post-opératoire montre une régression des hématomes, mais une lésion ischémique temporo-
occipitale gauche séquellaire de l’engagement temporal dans le territoire de l’artère cérébrale postérieure gauche, et une anomalie de la charnière cervico-occipitale avec sténose au niveau de C1, impression basilaire et une myélomalacie confirmant une instabilité de la charnière cervicale. L’examen neurologique reste normal sans conséquence de cette atteinte sur le plan sensitivo-moteur. Une immobilisation par collier cervical est mise en place. Sur le plan clinique, l’évolution neurologique est ensuite rassurante.
Sur le plan biologique, le bilan d’hémostase initial au diagnostic de ces hématomes sous-duraux montre un TP à 20 %, un facteur II inférieur à 10 %, un facteur V normal à 105 %, un facteur VII à 12 % et un facteur X à 11 %. Le dosage des facteurs de la voie endogène montre un facteur VIII à 176 %, un facteur IX à 23 % et un facteur XI à 80 %. Le facteur XIII est à 201 %.
Ce bilan est ainsi évocateur d’un déficit combiné en facteurs vitamine K-dépendants (II, VII, IX et X). Le patient va recevoir une supplémentation par du PPSB pour assurer le geste d’évacuation neurochirurgicale et l’hémostase post-
opératoire, puis une supplémentation régulière par de la vitamine K per os.
Par ailleurs, les explorations radiologiques du squelette confirment le syndrome de Binder par l’absence d’épine nasale, puis mettent en évidence une brachy-métaphalangie.
Elles confirment aussi la présence de calcifications multiples : trachéales, rachidiennes, du palais dur, du vomer, des cornets, du fémur. L’atteinte squelettique est ainsi évocatrice d’une forme sévère de chondrodysplasie ponctuée, c’est-à-dire une dysplasie osseuse congénitale caractérisée par la présence d’anomalies visibles sous forme de calcifications ponctuées au niveau des cartilages et visibles à la radiographie (Figure 1).
Dans le cadre de l’enquête étiologique de ce tableau clinique, une analyse pangénomique est réalisée et met en évidence une mutation homozygote du gène GGCX, au niveau de l’exon 2, classée comme pathogène (nomenclature c709TG, Leu70Arg). Les parents sont chacun hétérozygotes, asymptomatiques sur le plan clinique et leur bilan d’hémostase est normal.
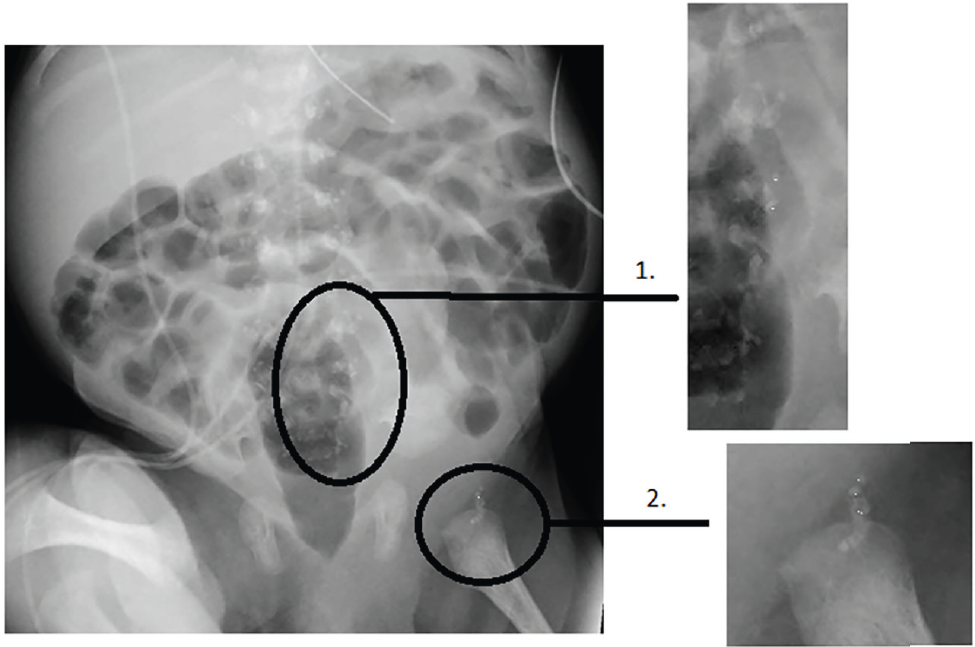
Figure 1 : Radiographie standard du patient – Chondrodysplasie ponctuée.
Figure 1: Standard X-ray of the patient – punctate chondrodysplasia.
1 : calcifi cations péri-vertébrales ; 2 : calcifi cations fémorales.
Actuellement âgé de 14 mois, ce jeune patient reçoit toujours une supplémentation orale par vitamine K à la posologie de 2 mg, un jour sur deux. Son dernier bilan d’hémostase montre un TP à 56 %, un facteur II à 36 %, un facteur V à 105 %, un facteur VII à 31 % et un facteur X à 47 %. Le TCK ratio est à 0,9. Le facteur IX est corrigé à 68 %. Sur le plan clinique, il ne présente aucune symptomatologie
hémorragique au quotidien, la polysomnographie est normale, l’examen neurologique est rassurant sans déficit moteur, il manipule les objets, tient assis, se déplace à 4 pattes, reproduit les gestes simples (fait « bravo » et
« coucou »), dit « maman ». La mobilité cervicale est normale, l’instabilité évolue favorablement et ne nécessite pour l’instant pas de geste neurochirurgical.
Discussion
Le gène GGCX est porté par le chromosome 2 (locus 2p12). La transmission des mutations se fait sur le mode autosomique récessif. Il code pour une enzyme GGCX ou Gamma Glutamyl Carboxylase, essentielle à la synthèse des facteurs de coagulation vitamine K-dépendants (facteurs II, VII, IX, X, protéines C et S). Cette enzyme permet une modification post-traductionnelle (rajout d’un groupe carboxyle) qui va permettre à ces facteurs de se fixer aux phospholipides et de jouer leur rôle dans l’hémostase. La vitamine K est un cofacteur essentiel à cette réaction de
carboxylation (Figure 2). Ce jeune patient a d’ailleurs également un déficit modéré en protéines C et S associé. Bien que le rôle principal de GGCX soit lié à la coagulation, cette enzyme est également impliquée dans d’autres processus physiologiques où d’autres protéines dépendantes de la carboxylation sont nécessaires, et notamment la régulation du métabolisme osseux (1).
Sur le plan clinique, un déficit enzymatique en GGCX va ainsi se traduire par une synthèse défectueuse des facteurs de la coagulation vitamine-K dépendants associée à une atteinte osseuse, de sévérité variable, allant d’un phénotype de
Binder isolé à des anomalies squelettiques plus étendues de type chondrodysplasie ponctuée.
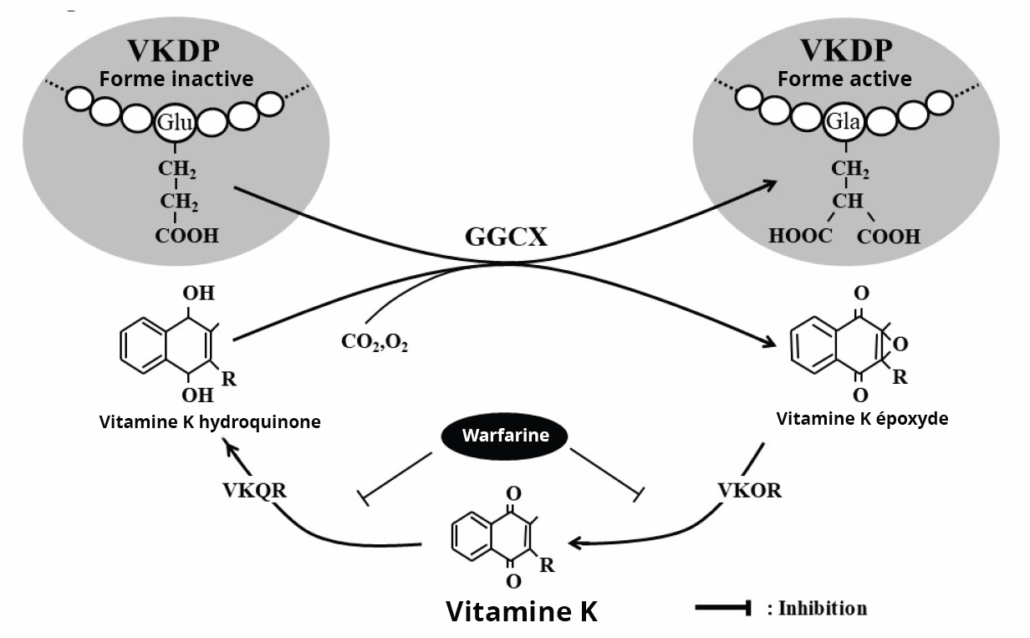
Figure 2 : Boucle enzymatique de la carboxylation des facteurs vitamine K-dépendants (1).
Figure 2: Enzymatic loop of carboxylation of vitamin K-dependent factors (1).
Certains variants de GGCX sont associés à une diminution significative de la carboxylation des facteurs de coagulation, corrélé à un phénotype hémorragique plus sévère (2). Il existe donc aussi une variabilité d’expression du phénotype hémorragique (3) allant des hémorragies périnatales sévères au diagnostic fortuit à l’âge adulte, y compris au sein de fratries porteuses des mêmes mutations (4).
Sur le plan thérapeutique, les patients sont traités par une supplémentation avec de la vitamine K, avec une réponse variable, probablement liée à la sévérité du déficit enzymatique (5). Il est d’ailleurs possible qu’un déficit complet en GGCX soit parfois létal au stade embryonnaire (6).
Par ailleurs, il est important de noter que ce tableau clinique mime l’embryofœtopathie liée à la prise d’antivitamines K durant la grossesse. La vitamine K passe en effet la barrière fœto-placentaire, mais sa concentration plasmatique fœtale reste faible. En effet, l’immaturité métabolique hépatique du fœtus limite le stockage des vitamines liposolubles et ne permet qu’une faible synthèse des facteurs de la coagulation vitamine-K dépendants (7). Néanmoins, les manifestations cliniques hémorragiques de ces déficits sont généralement décrites après la naissance. L’hypothèse émise est celle que la vitamine K maternelle permettrait chez ces fœtus déficitaires en GGCX une synthèse minimale de facteurs vitamine K-dépendants qui les protégerait du risque d’hémorragie anténatale, néanmoins déjà une fois décrit dans la littérature (8).
Enfin, il existe des diagnostics différentiels à cette pathologie rare (4). Les déficits combinés en facteurs de coagulation vitamine-K dépendants peuvent aussi être liés à des mutations du gène VKORC1, de transmission autosomique récessive également. Le phénotype est similaire associant un déficit en facteurs de coagulation, un syndrome hémorragique clinique et une atteinte osseuse : hypoplasie nasale et ponctuation épiphysaire (9,10).
Un autre diagnostic différentiel est le syndrome de Keutel, lié à des mutations homozygotes du gène MGP codant pour la protéine MGP (Matrix Gla Protein), qui est impliquée dans le processus de minéralisation osseuse, et inhibe également la calcification des vaisseaux et du cartilage. Sur le plan clinique, le phénotype osseux est donc ressemblant : calcifications anormales des cartilages avec ponctuations épiphysaires, syndrome de Binder, brachy-téléphalangie, mais aussi sténoses pulmonaires, communication interventriculaire, et surdité (11). L’activité de la protéine MGP est vitamine K-dépendante, et nécessite qu’elle soit carboxylée, mais il n’y a pas d’anomalie de l’hémostase associée dans ce syndrome.
D’autre part, il existe aussi des chondrodysplasies ponctuées liées à l’X, dues à une mutation du gène ARSE, codant pour l’arylsulfatase E. Le tableau clinique est celui d’une atteinte osseuse sans anomalie de l’hémostase associée (12).
Un dernier point important concerne le conseil génétique. Le diagnostic posé chez notre patient d’une pathologie due à une mutation homozygote de GGCX est essentiel pour le conseil génétique, car la transmission est autosomique récessive. Ceci implique un risque de récurrence de 25 % à chaque grossesse ultérieure. Il est donc important d’en informer les parents, de discuter des possibilités de diagnostic prénatal voire pré-implantatoire, d’assurer un suivi échographique rapproché des grossesses ultérieures, mais aussi d’évoquer la variabilité des phénotypes possibles, plus ou moins sévères. Sur le plan thérapeutique, il est également possible de proposer une supplémentation maternelle en vitamine K au cours de la grossesse. Concernant le nouveau-né à naître, en l’absence de diagnostic prénatal, un dosage des facteurs vitamine K-dépendants peut être réalisé au sang de cordon, et une supplémentation orale en vitamine K doit être initiée dans l’attente de la confirmation diagnostique éventuelle.
Conclusion
Ce cas illustre la complexité clinique et diagnostique des syndromes associés aux maladies osseuses constitutionnelles. La mutation homozygote du gène GGCX identifiée chez ce patient permet d’expliquer l’ensemble du phénotype et d’initier une prise en charge adaptée, notamment par une supplémentation en vitamine K. Ce diagnostic génétique revêt une importance particulière, non seulement pour l’orientation thérapeutique, mais aussi pour le conseil génétique et la mise en place de mesures préventives lors de futures grossesses. Il souligne également la nécessité d’une vigilance accrue face à ces tableaux cliniques rares, pour éviter un retard de diagnostic aux conséquences potentiellement graves.
• Mutation du gène GGCX responsable d’un défi cit combiné en facteurs de coagulation vitamine K-dépendants et d’anomalies squelettiques de type chondrodysplasie ponctuée, avec expression phénotypique variable.
• Présentation néonatale complexe : syndrome de Binder détecté en anténatal, suspicion de chondrodysplasie, puis hémorragie intracrânienne sévère révélant le défi cit hémostatique.
• Importance du diagnostic génétique : permet d’adapter la prise en charge, de confi rmer l’étiologie, et d’initier un conseil génétique pour les familles.
• Diagnostics différentiels à évoquer : embryofoetopathies aux AVK et mutations du gène VKORC1.
Liens d’intérêts : l’auteur déclare ne pas avoir de lien d’intérêt en rapport avec cet article.