RÉSUMÉ
La drépanocytose est la maladie monogénique la plus fréquente en France, touchant plus de 30 000 personnes. Elle associe trois grandes catégories de manifestations cliniques avec une grande variabilité d’expression selon les individus atteints : une anémie hémolytique chronique avec épisodes d’aggravation aiguë, une susceptibilité aux infections bactériennes et des phénomènes vaso-occlusifs. Il existe chez les patients drépanocytaires un terrain thrombo-inflammatoire qui se traduit par une augmentation de l’incidence des thromboses artérielles et veineuses pouvant aller jusqu’à 25 % chez les patients avant 40 ans. Ce risque thromboembolique accru est sous-tendu par des mécanismes physiopathologiques variables et intriqués : l’exposition augmentée de la phosphatidylsérine et des phospholipides anioniques à la surface des globules rouges, l’augmentation de la génération de thrombine par l’adsorption des protéines C et S, la production accrue de NETs, la thrombocytose chronique, et enfin l’augmentation de la génération de microparticules qui participent à la génération de thrombine. Il n’y a pas de recommandations spécifiques sur la prise en charge des événements thromboemboliques veineux chez les patients drépanocytaires. L’anticoagulation préventive par HBPM doit être systématique chez les patients pubères et alités et doit être largement évoquée chez l’enfant en fonction du risque thrombotique quel que soit l’âge. À visée curative, les AOD constituent une thérapeutique de choix, du fait de leur efficacité mais également d’une action anti-inflammatoire bénéfique dans ce contexte.MOTS CLÉS
drépanocytose, NETose, thrombo-inflammation, thromboembolie veineuse
ABSTRACT
Sickle cell disease (SCD) is the most common monogenic disorder in France, affecting more than 30,000 individuals. It encompasses three major categories of clinical manifestations, with considerable variability in expression among affected individuals: chronic hemolytic anemia with episodes of acute exacerbation, increased susceptibility to bacterial infections, and vaso-occlusive phenomena. Patients with SCD exhibit a thrombo-inflammatory state, reflected by an increased incidence of arterial and venous thrombosis, reaching up to 25% before the age of 40. This heightened thromboembolic risk is driven by multiple, interrelated pathophysiological mechanisms: increased exposure of phosphatidylserine and anionic phospholipids on the red blood cell surface; enhanced thrombin generation through adsorption of proteins C and S; increased production of neutrophil extracellular traps (NETs); chronic thrombocytosis; and increased generation of microparticles contributing to thrombin formation. There are no specific guidelines for the management of venous thromboembolic events in patients with SCD. Preventive anticoagulation with low-molecular-weight heparin (LMWH) should be systematic for pubertal and bedridden patients and should be strongly considered for children based on thrombotic risk, regardless of age. For curative treatment, direct oral anticoagulants (DOACs) represent a preferred therapeutic option, owing not only to their efficacy but also to their beneficial anti-inflammatory effects in this context.
KEYWORDS
NETose, sickle cell disease, thromboinflammation, venous thromboembolism
INTRODUCTION
La drépanocytose est une affection héréditaire de l’hémoglobine qui touche près de 8 millions de personnes dans le monde. En France, il s’agit de la maladie monogénique la plus fréquente, touchant plus de 30 000 personnes, avec environ 600 nouveaux cas dépistés chaque année. Il s’agit d’une maladie sévère, ayant un impact majeur sur la qualité et l’espérance de vie des patients, alors même que les options thérapeutiques disponibles demeurent très limitées.
Les syndromes drépanocytaires majeurs résultent d’une mutation ponctuelle du gène codant la chaîne bêta de l’hémoglobine, conduisant à la production d’une hémoglobine anormale (HbS). En conditions désoxygénées, l’HbS a tendance à polymériser, ce qui modifie profondément les propriétés des globules rouges (GR) : perte de la déformabilité, fragilité membranaire conduisant à l’hémolyse, et adhérence anormale des GR à l’endothélium vasculaire ainsi qu’aux autres cellules sanguines.
La drépanocytose associe trois grandes catégories de manifestations cliniques avec une grande variabilité d’expression selon les individus atteints :
• une anémie hémolytique chronique avec épisodes d’aggravation aiguë ;
• une susceptibilité aux infections bactériennes ;
• des phénomènes vaso-occlusifs.
Les crises vaso-occlusives (CVO) résultent de l’obstruction des capillaires artériolo-veineux par les globules rouges falciformés, les neutrophiles et les plaquettes activées. Elles se manifestent par des douleurs aiguës, extrêmement intenses, imprévisibles, le plus souvent localisées aux os mais pouvant toucher tous les organes. Parmi les manifestations vaso-occlusives les plus graves figure le syndrome thoracique aigu (STA) caractérisé par l’obstruction des capillaires de la microcirculation pulmonaire, générant une détresse respiratoire aiguë grave. Les CVO constituent le motif le plus fréquent d’hospitalisation, altèrent profondément la qualité de vie des patients et contribuent à terme à l’altération de tous les organes (reins, foie, poumon, coeur, peau, rétine…) et à la morbi-mortalité de la maladie.
DRÉPANOCYTOSE ET THROMBOSE
Les patients atteints de drépanocytose présentent un risque accru de thrombose à la fois veineuse et artérielle. Les événements thromboemboliques veineux (ETEV), incluant thrombose veineuse profonde et embolie pulmonaire, sont fréquents, avec une incidence cumulée pouvant aller jusqu’à 25 % chez les patients avant 40 ans (1). Le STA s’accompagne notamment fréquemment d’une microthrombose pulmonaire, parfois difficile à distinguer d’une embolie pulmonaire classique.
Physiopathologie du risque thrombotique dans la drépanocytose
Les mécanismes physiopathologiques conduisant à ce risque thrombotique dans la drépanocytose sont complexes et imbriqués.
Les GR drépanocytaires présentent une exposition accrue de la phosphatidylsérine (PS) à la membrane, favorisant ainsi la génération de thrombine ; les drépanocytes polymérisés présentent également une exposition plus importante de phospholipides (PL) anioniques à leur surface en comparaison aux hématies saines. Il résulte de ces mécanismes une diminution de la protéine S par interaction avec les PL exposés, de la protéine C, ainsi qu’une augmentation du facteur tissulaire (Figure 1).
Les GR, les plaquettes et les polynucléaires neutrophiles (PNN) des patients drépanocytaires présentent tous une adhérence accrue et délétère à l’endothélium vasculaire, en raison notamment de l’expression anormale de molécules d’adhésion à la surface des GR, des Intégrines MAC-1 et LFA-1 ou encore du couple P-Selectine/PSLG1 à la surface des cellules endothéliales, des plaquettes et des PNN, favorisant la stase et la coagulation (Figure 1) (2).
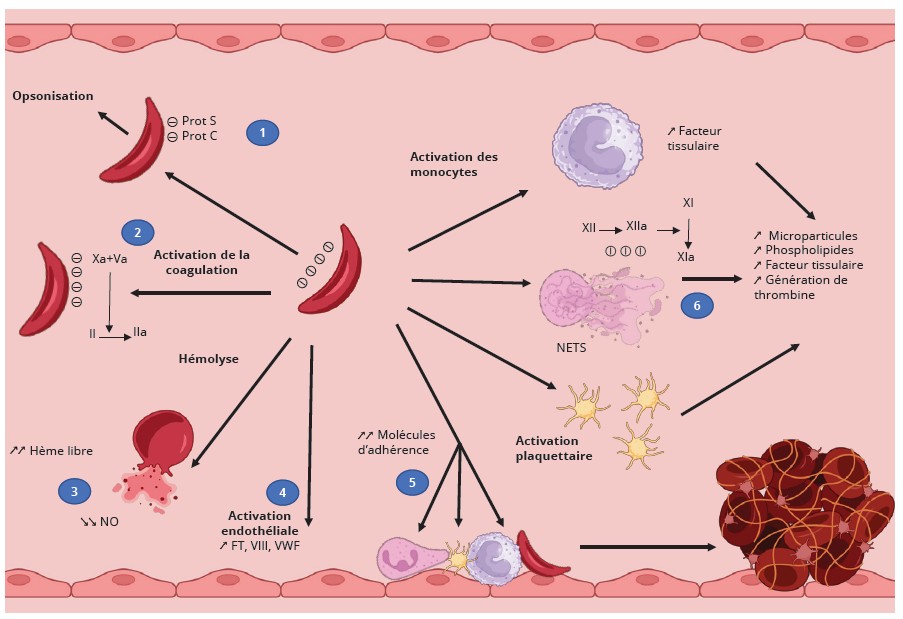
Figure 1 : Mécanismes physiopathologiques de la thrombo-inflammation dans la drépanocytose.
Figure 1: Pathophysiological mechanisms of thromboinflammation in sickle cell disease.
1 : exposition accrue de phospholipides anioniques (-) à la surface des globules rouges drépanocytaires responsables d’une diminution de la protéine S, de la protéine C, et d’une augmentation du facteur tissulaire ; 2 : génération de thrombine (IIa) à la surface des drépanocytes exprimant les phospholipides anioniques (phosphatidyl-sérine) ; 3 : libération de l’arginase érythrocytaire par l’hémolyse chronique, diminuant la synthèse endothéliale de NO et entraînant une vasoconstriction locale. 4 : activation des cellules endothéliales par les
cellules sanguines circulantes, responsable d’une augmentation du facteur tissulaire (FT), du facteur VIII et du facteur Willebrand ; 5 : augmentation de l’adhérence des polynucléaires neutrophiles, des plaquettes et des érythrocytes, participant à la stase cellulaire et à l’obstruction veineuse ; 6 : production accrue de NETs, responsable d’une activation du système contact ainsi que du facteur XII aboutissant à la génération de thrombine. L’activation conjointe des monocytes et des plaquettes favorise la libération de facteur tissulaire ainsi que la production de microparticules procoagulantes.
De fait, bien que longtemps considérée comme une maladie touchant uniquement le GR, la drépanocytose apparaît aujourd’hui comme une véritable maladie inflammatoire chronique. Pour des raisons physiopathologiques encore mal connues, les patients drépanocytaires présentent à l’état basal une hyperleucocytose à PNN et à monocytes, avec des anomalies fonctionnelles et phénotypiques de ces cellules. Parmi ces anomalies fonctionnelles, la production accrue de NETs (Neutrophil Extracellular Traps) joue un rôle majeur dans la physiopathologie de la drépanocytose, en particulier dans les phénomènes inflammatoires, vaso-occlusifs et prothrombotiques. Les NETs piègent les GR falciformes, les plaquettes et les leucocytes, augmentent l’adhérence cellulaire à l’endothélium et favorisent la formation de microthrombi par un effet procoagulant via l’exposition de phospholipides anioniques et une activation du système contact par le facteur XII, entraînant une obstruction des microvaisseaux et une ischémie tissulaire (3).
Enfin, l’hémolyse chronique libère l’arginase érythrocytaire, qui entre en compétition avec la NO synthase, diminuant ainsi la synthèse endothéliale de NO et entraînant une vasoconstriction locale (Figure 1).
Les plaquettes sont également probablement un autre acteur clé des phénomènes thrombotiques dans la drépanocytose. En effet, les patients présentent une thrombocytose modérée mais chronique, dont l’origine reste mal comprise, avec une activation basale de l’inflammasome plaquettaire. Chez les patients drépanocytaires, les plaquettes libèrent ainsi une grande quantité de thrombospondine-1 et de CD40L, deux molécules impliquées dans le STA. Leur adhérence aux neutrophiles et aux cellules endothéliales est accrue et inappropriée, notamment via la P-sélectine et l’intégrine αIIbβ3, menant ainsi à l’expression endothéliale d’ICAM1, de E-sélectine et d’IL8. De plus, l’activation plaquettaire favorise la formation d’agrégats GR-neutrophiles, jouant un rôle critique dans l’initialisation et la propagation des CVOs (4).
Les microparticules (MP) circulantes, vésicules intactes provenant des membranes cellulaires, sous l’action de différents facteurs comme l’inflammation, le stress oxydatif ou la sécrétion de cytokines, pourraient également participer au phénotype prothrombotique. Dans la drépanocytose, les MP circulantes proviennent des hématies, des plaquettes, des polynucléaires neutrophiles et des cellules endothéliales : elles augmentent l’exposition à la PS ainsi qu’au facteur tissulaire et participent ainsi à la génération de thrombine (5).
Enfin, le système du complément est un dernier acteur pouvant concourir au profil prothrombotique de ces patients. En effet, thrombose et activation de la voie du complément sont étroitement liées par des mécanismes bidirectionnels formant une immunothrombose, où l’immunité innée et l’hémostase s’auto-entretiennent. L’activation du complément, quelle que soit la voie (classique, lectines ou alterne), génère des fragments effecteurs comme C3a et C5a. Ces anaphylatoxines activent les cellules endothéliales, les plaquettes et les leucocytes, induisant une expression accrue du facteur tissulaire (FT), une augmentation de l’adhérence cellulaire et une perte des propriétés anticoagulantes de l’endothélium. C5a est particulièrement puissant pour activer les neutrophiles et favoriser la libération de NETs, qui constituent une surface procoagulante et un support à la formation de thrombus. Le complexe d’attaque membranaire C5b-9, même à des concentrations sub-lytiques, active l’endothélium et les plaquettes. Il favorise la libération de microparticules riches en FT et augmente l’exposition de phospholipides procoagulants, renforçant ainsi la génération de thrombine. Inversement, la coagulation active à son tour le complément. La thrombine, le facteur Xa et la plasmine peuvent cliver directement C3 et C5, indépendamment des convertases classiques, amplifiant la cascade du complément. Les plaquettes activées lient et concentrent les protéines du complément à leur surface, facilitant leur activation locale au sein du thrombus (Figure 1) (6).
PRISE EN CHARGE
Pistes thérapeutiques antithrombotiques en contexte de drépanocytose
Actuellement, il n’y a pas de recommandations officielles de prophylaxie systématique des ETEV chez les drépanocytaires. Néanmoins, certaines études se sont penchées sur cette problématique : une étude rétrospective publiée en 2023 portant sur 7 202 patients drépanocytaires de 13 à 21 ans montre une augmentation de la prophylaxie de 1,3 % des admissions en 2010 à 14,4 % des admissions en 2021 avec une augmentation croissante des anticoagulants oraux directs (AOD) (7).
Le protocole national de diagnostic et de soins (PNDS) pour les syndromes drépanocytaires majeurs recommande une anticoagulation préventive par HBPM systématique pour les patients pubères et alités. Avant la puberté, elle peut être discutée chez les patients à risque thrombotique (antécédent d’ETEV, obésité, génotype SC, porteurs d’un cathéter central, COVID-19) quel que soit l’âge.
Parmi les molécules fréquemment utilisées, on retrouve :
• les héparines de bas poids moléculaire (HPBM) : en plus d’une efficacité démontrée dans la prise en charge des ETEV, des études montrent une diminution de la douleur et une diminution de la durée d’hospitalisation en cas d’utilisation prophylactique au cours d’une crise vasoocclusive (8) ;
• les AOD : plusieurs études montrent une efficacité similaire entre AVK et AOD pour le traitement et la prévention des récidives de thromboses, cependant les AOD seraient à privilégier du fait d’une diminution des complications hémorragiques et de moindres variations inter et intra individuelles. Étant contre-indiqués pendant la grossesse et l’allaitement, les HBPM restent le traitement de choix dans ce cas. De plus, un nombre croissant d’études montre une action anti-inflammatoire des AOD avec notamment une diminution des marqueurs d’inflammation comme l’IL-6 et le TNF-α, les rendant particulièrement intéressants dans la drépanocytose (9) ;
• les anti-agrégants plaquettaires : les études récentes ne montrent pas de bénéfice clinique chez les patients drépanocytaires. De plus, en raison de la thrombocytose fréquente de ces patients, une diminution acquise de l’activité du facteur Willebrand peut être observée et majorer le risque d’hémorragies cutanéomuqueuses(10).
Différentes perspectives sont à l’étude pour permettre une prévention au long cours du risque thrombotique chez les patients drépanocytaires, parmi lesquelles :
• les anti-XI : actuellement à l’étude dans diverses indications comme la fibrillation atriale, leur application dans la prévention des thromboses à la fois artérielles et veineuses au cours de la drépanocytose pourrait être prometteuse. Du fait du rôle du facteur XI et plus globalement du système contact (FXII, kininogène et prékallicréine) dans le phénotype procoagulant des patients, son inhibition pourrait diminuer l’hypercoagulabilité tout en étant associée à un risque hémorragique moindre ;
• les inhibiteurs du complément tels que l’éculizumab ont rapporté dans certains cas un effet bénéfique en cas de syndrome d’hyperhémolyse post-transfusionnelle, et pourraient prévenir la survenue de complications thrombotiques dans ces situations (11) ;
• les héparines non coagulantes sulfatées (S-NACH),dérivés d’HBPM sans effet anticoagulant par ajout d’un groupement sulfate. Ces molécules présentent un effet d’inhibition de la liaison de la P-selectine, ce qui pourrait diminuer l’adhérence des GR à l’endothélium et réguler l’inflammation en jouant sur les niveaux de cytokines (12) ;
• le sevuparin est une molécule dérivée des héparines qui inhibe l’adhérence des GR et des leucocytes à l’endothélium via la p-selectine et la l-selectine (13) ;
• enfin, l’isoquercétine est un flavonoïde dont le rôle a été étudié dans la prévention des thromboses dans le cancer. Elle fait l’objet d’essais cliniques dans la drépanocytose en prévention du risque thrombotique et semble montrer une diminution de certains biomarqueurs thrombo-inflammatoires et de l’agrégation plaquettaire chez ces patients (14).
CONCLUSION
La drépanocytose est une pathologie inflammatoirechronique majorant le risque thrombotique veineux etartériel, par le biais de nombreux mécanismes procoagulantscomplexes et imbriqués. La prévention et le traitement de ces thromboses sont indispensables, et doivent être adaptés au contexte clinique et aux facteurs derisque des patients. Les AOD constituent une thérapeutiquede choix, du fait de leur efficacité mais également d’uneaction anti-inflammatoire bénéfique dans ce contexte.
POINTS CLÉS À RETENIR
• Les événements thromboemboliques veineux sont fréquents chez les patients drépanocytaires et doivent être systématiquement évoqués, y compris chez l’enfant avant la puberté.
• L’anticoagulation préventive par HBPM doit être systématique pour les patients alités, pour tous après la puberté et chez l’enfant en cas de majoration du risque thrombotique (antécédent d’ETEV, obésité, génotype SC, porteurs d’un cathéter central, COVID-19).
• En traitement chez l’adulte drépanocytaire, les AOD constituent une thérapeutique de choix, du fait d’une action anti-inflammatoire bénéfique dans ce contexte.
Liens d’intérêts : les auteurs déclarent ne pas avoir de lien d’intérêt en rapport avec cet article.