RÉSUMÉ
Le facteur XI de la coagulation est un zymogène. Sa structure sous-tend deux mécanismes d’activation (par le facteur XIIa – système contact – ou la thrombine – rétrocontrôle positif), et de multiples fonctions, en plus de l’activation du facteur IX. Ce facteur articule le système contact avec la ténase intrinsèque (voie intrinsèque de la coagulation). Parce qu’il est activable par la thrombine, il peut jouer un rôle dans l’hémostase physiologique démarrée par le facteur tissulaire (FT). Cette implication du facteur XI dans la coagulation n’est pas explorée par le temps (de coagulation) céphaline avec activateur (TC + A). Il faut utiliser le FT à très faibles concentrations, et travailler au mieux en présence de plaquettes, ce qui complique fortement la démarche. Le facteur IX peut être aussi activé par la kallicréine, court-circuitant ainsi les facteurs XII et XI, et cette voie courte pourrait expliquer en partie la variabilité du phénotype clinique (hémorragique) des déficits constitutionnels en facteur XI. Le degré d’implication du facteur XI dans l’hémostase pour un individu à un moment donné n’est pas prévisible par les explorations menées en laboratoire clinique. L’intervention du facteur XI dans les thromboses semble dépendre de l’implication des polyphosphates et de leur longueur de chaîne, et de la formation de pièges extracellulaires des neutrophiles ou NETs. L’activation délétère de la coagulation au contact de surfaces « étrangères » passe par les facteurs XII et XI. L’activation du système contact et du facteur XI dès le prélèvement de sang constitue un véritable poison pour l’étude fine de la coagulation.MOTS CLÉS
coagulation, facteur XI, surfaces étrangères, système contact, thrombose
ABSTRACT
The structure of the zymogen coagulation factor XI enables two activation mechanisms (by factor XIIa – contact system – or thrombin – positive feedback), and activation of factor IX and other functions, some beyond coagulation. Factor XI links the contact system with intrinsic tenase (i.e. intrinsic coagulation pathway). Because it can be activated by thrombin, it plays a role in physiological haemostasis initiated by tissue factor (TF). Such an involvement is not explored by the aPTT clotting time. TF must be used in very low concentrations instead and one should work in the presence of platelets, which greatly complicates the study procedure. Factor IX can also be activated by kallikrein, thus bypassing factors XII and XI, and this shortcut could partly explain the variability of the clinical (haemorrhagic) phenotype of inherited factor XI deficiencies. The degree of the involvement of factor XI in haemostasis for an individual at a given time cannot be predicted by the available clinical laboratory tests. The role of factor XI in thrombosis appears to depend on the involvement of polyphosphates and on their chain length, and on the formation of extracellular neutrophil traps (NETs). The harmful activation of coagulation upon contact with “foreign” surfaces involves factors XII and XI. The activation of the contact system and factor XI during blood collection makes the refined study of coagulation highly tricky.
KEYWORDS
coagulation, contact system, factor XI, foreign surfaces, thrombosis
INTRODUCTION
Le facteur XI de la coagulation a été identifié pour la première fois en 1953 par Robert Rosenthal et ses collaborateurs comme le constituant plasmatique manquant chez des membres d’une famille avec un syndrome hémorragique modeste, en rapport avec des traumatismes (1).
Sa structure et ses multiples fonctions le distinguent des autres facteurs de la coagulation. Il fait l’objet d’une attention accrue car, ces dernières années, il est la cible de nouveaux médicaments anticoagulants en développement (2), dans l’espoir d’accroître le différentiel entre affaiblissement de l’hémostase (effet pro-hémorragique) et protection contre la thrombose (3). Ce facteur joue un rôle dans l’hémostase (principalement, mais non exclusivement, en activant le facteur IX), mais ce rôle est nettement plus modeste que celui des facteurs anti-hémophiliques (4). Le facteur XI(a) a donc été considéré comme une bonne cible pour l’anticoagulation, son inhibition pouvant faire courir un risque hémorragique plus faible que l’inhibition du facteur Xa, quoique non nul. Toutefois, il existe une réserve majeure : en cas de déficit constitutionnel, le risque hémorragique, qui n’est pas nul, est très mal prédit par le niveau plasmatique résiduel du facteur (5,6) qui suggère une grande variabilité interindividuelle, mal expliquée, de l’implication du facteur XI dans la coagulation. De plus, il faut répondre à l’interrogation suivante : comment concevoir – et expliquer – que le facteur XI, amplificateur de la génération de thrombine, joue un rôle important dans les thromboses alors qu’il semble plus modeste pour l’hémostase physiologique ?
PRESENTATION DU FACTEUR XI (4,7)
Le facteur XI est une glycoprotéine de masse moléculaire 160 KDa, synthétisée par les hépatocytes. Son gène (F11) est situé en 4q35 et résulte d’une duplication de celui (Klkb1) de la prékallicréine (PK), avec laquelle il présente logiquement des homologies de structure ; XI et PK sont des paralogs. Mais le facteur XI a évolué passant d’un composant du système « KKS » (système plasmatique kallicréine – kinine, mettant l’accent sur sa fonction inflammatoire) à un zymogène de proté(in)ase de la coagulation.
C’est le seul facteur de coagulation qui soit un homodimère (d’où sa masse moléculaire la plus élevée parmi les facteurs de la coagulation). Les domaines apple 4 de chaque monomère sont adossés l’un à l’autre, avec interactions hydrophobes, ponts salins, et un pont disulfure (liaison covalente impliquant Cys-321). Un monomère comprend 607 résidus d’acides aminés et est constitué d’un domaine catalytique (avec la triade catalytique des sérines-protéases) et de quatre domaines dits apple : le premier repose sur les derniers comme une tasse sur une sous-tasse (cup-and-saucer).
Les interactions fondamentales se font avec le kininogène de haute masse moléculaire (KH’P’M), le facteur XIIa, le facteur IIa (thrombine), les substrats – dont le facteur IX – et les inhibiteurs physiologiques. Elles impliquent tous les domaines, de manière propre à chaque interactant, avec notamment deux sites de liaison aux molécules polyanioniques (anion binding sites ou ABS), et notamment aux polyphosphates et aux glycosaminoglycanes. L’interactome du facteur XI(a) est schématisé dans la figure 1. Nous discuterons plus loin certaines de ces interactions.
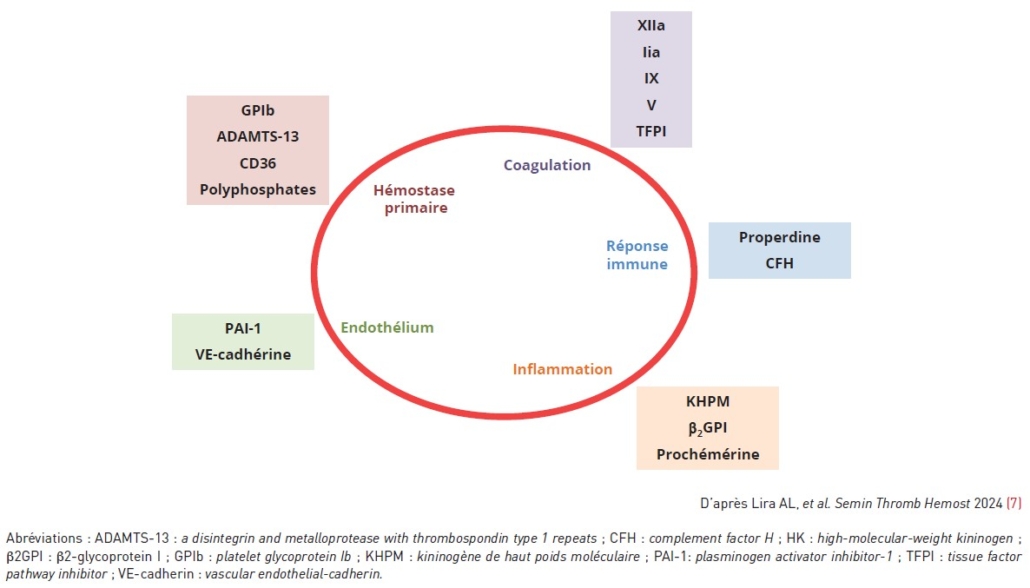
Figure 1 : L’interactome du facteur XI(a).
Figure 1: The factor XI(a) interactome.
Les (nombreux) inhibiteurs physiologiques du facteur XIa ne sont pas indiqués.
Le facteur XI circule dans le plasma associé presque en totalité au KHPM de manière non covalente à la concentration d’environ 30 nM (15-45 nM), exprimée en pourcentage ou en U/dL : 50 à 150. L’immobilisation du facteur XI sur une surface étrangère se ferait surtout par l’intermédiaire du KHPM.
Le facteur XI est placé à l’interface de la phase contact (9) et de la ténase intrinsèque (facteurs anti-hémophiliques) : il active le facteur IX, propriété qu’il partage avec le facteur VIIa en présence de facteur tissulaire (FT) ; c’est la boucle de Josso (Figure 2).
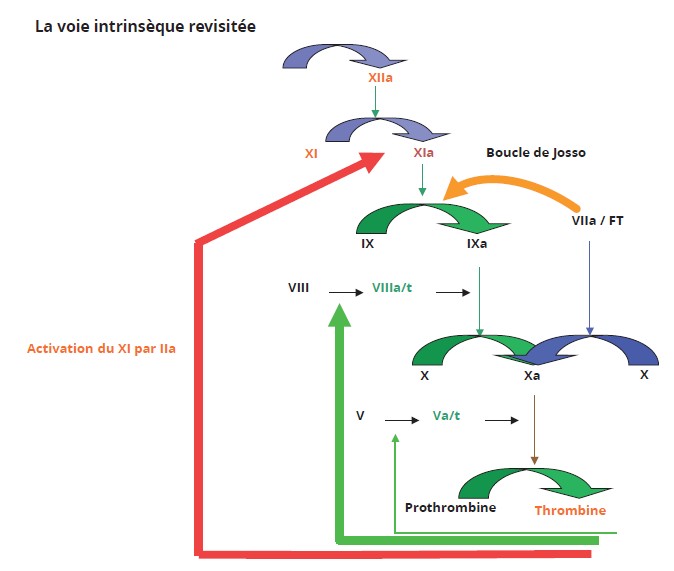
Figure 2 : Les boucles de rétrocontrôle positif de la thrombine – implication du facteur XI dans l’hémostase physiologique.
Figure 2: Positive feedback loops of thrombin – involvement of factor XI in physiological hemostasis.
Le facteur XI joue un rôle dans l’hémostase physiologique, le facteur XII non. La voie intrinsèque à partir du facteur XI est impliquée suite à l’exposition au FT du fait du rétrocontrôle positif par les premières traces de thrombine. La boucle de Josso fait que la ténase intrinsèque est aussi mise en jeu. Flèches vertes : activation des facteurs V et VIII par la thrombine.
Il partage avec les trois protéines du système contact (le facteur XII, la prékallicréine ou PK, zymogène de la kallicréine, et le KHPM), une synthèse hépatocytaire, mais aussi l’absence de domaine Gla (et donc la vitamine K n’exerce aucun rôle lors de cette synthèse). Son fonctionnement ne nécessite pas de calcium, qui peut donc être mis en jeu en milieu citraté (10) (Figure 3).
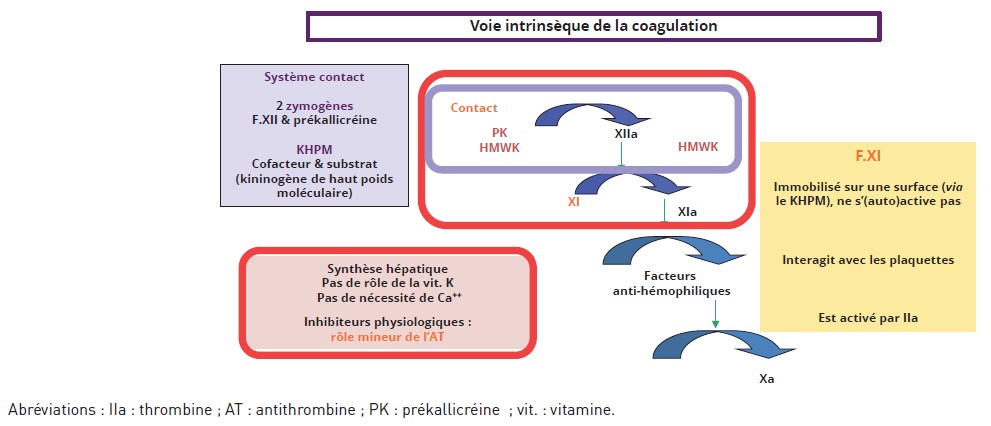
Figure 3 : Le facteur XI articule système contact et ténase intrinsèque.
Figure 3: Factor XI links the contact system and intrinsic tenase.
Similitudes et différences entre le facteur XI et les deux autres zymogènes – le KHPM (ou HMWK), un cofacteur, et le substrat de la kallicréine (avec libération de kinine).
C’est une propriété intrinsèque au sang (au plasma) que de coaguler au contact d’une surface « étrangère » (exogène), car elle ne fait appel à aucun acteur biologique de l’organisme qui ne soit pas présent dans le sang (extrinsèque), ce qui est exploré avec l’activateur utilisé pour le temps de coagulation (temps de céphaline avec activateur -aPTT en anglais) ; à noter qu’il convient donc de parler de voie intrinsèque, et non pas « endogène » (Figure 4). Cette propriété serait utile pour les animaux terrestres, qui se blessent fréquemment sur le sol (riche en silicates) du fait de leur mode de déplacement (rampant, quadrupède…). Elle ne l’est plus pour les cétacés (mammifères retournés en milieu aquatique), qui se sont « débarrassés » du facteur XII ; pour les êtres humains (puisqu’erectus), elle l’est beaucoup moins, du moins pour une hémostase physiologique (8). Le système contact est une « sentinelle », qui, en présence de polyphosphates à longues chaînes (non-soi), ou de polysaccharides non physiologiques (agents infectieux), contribue à l’inflammation et à la mise en jeu de l’immunité innée (9).
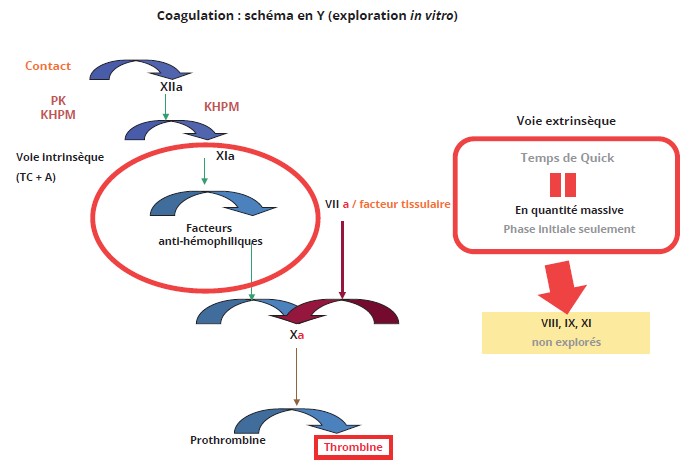
Figure 4 : La coagulation telle qu’explorée en médecine de laboratoire.
Figure 4: Coagulation as explored in laboratory medicine.
Deux temps de coagulation sont utilisés : le TC + A (temps de céphaline avec activateur) explore la voie intrinsèque qui débute par la phase contact, laquelle est connectée à la ténase intrinsèque par le facteur XI ; le temps de Quick explore la coagulation par la voie extrinsèque dans laquelle le facteur XI ne joue aucun rôle, du fait des quantités massives de facteur tissulaire ajoutées.
ACTIVATION ET INHIBITION PHYSIOLOGIQUES DU FACTEUR XI
La coupure d’une liaison peptidique au sein du facteur XI par le facteur XIIa produit deux chaînes, qui restent reliées par des ponts disulfure (11) ; la chaîne légère correspond au domaine catalytique, qui devient alors actif. L’activation est aussi possible par la thrombine (12). Ce double mode d’activation règle une question qui a longtemps intrigué les hémostasiens : alors que les facteurs XII et XI sont tous deux explorés par le TC + A, pourquoi le déficit de ces deux protéines a-t-il un impact différent sur l’hémostase permettant l’arrêt d’un saignement ? Le déroulement de l’hémostase physiologique débuté par la mise en contact du sang avec le FT (voie extrinsèque) exposé en cas de brèche vasculaire est dans ses grandes lignes le suivant (13) : le complexe VIIa-FT enclenche des réactions peu efficaces qui aboutissent à la formation de traces de (meizo) thrombine via le facteur Xa. Plus lentement, le facteur VIIa lié au FT active le facteur IX. Les premières traces de thrombine sont suffisantes pour la mise en jeu des boucles d’amplification : d’une part l’activation plaquettaire avec l’exposition de phospholipides anioniques permettant l’assemblage des complexes formant la ténase intrinsèque et la prothrombinase ; d’autre part l’activation des facteurs VIII et V, les cofacteurs respectifs (3). Enfin, de plus grandes quantités de thrombine permettent aussi l’activation du facteur XI, le zymogène des rétrocontrôles positifs, contrairement aux deux cofacteurs (V et VIII). Les éléments du système KKS ne jouent là aucun rôle et par conséquent leur déficit n’est logiquement pas hémorragipare.
Cependant, et ce fait est important, le facteur IX peut être activé par la kallicréine, court-circuitant ainsi les facteurs XII et XI (14) ; c’est même la voie d’activation ancestrale du facteur IX, apparue tôt dans l’évolution des organismes pluricellulaires, avant l’apparition du facteur XI par duplication du gène Klkb1 de la PK. L’évolution du gène F11 a rendu son produit plus efficace pour activer le facteur IX (8). La voie courte kallicréine – facteur IX, selon qu’elle est plus ou moins active, pour des raisons qui restent à élucider pleinement, pourrait expliquer au moins en partie la variabilité du phénotype clinique (hémorragique) des déficits constitutionnels en facteur XI. Elle constituerait aussi une limite majeure aux approches anticoagulantes ciblant le facteur XI(a) mais aussi le facteur XII(a), dès lors que de la kallicréine formée jouerait un rôle dans la thrombogenèse.
Le facteur XI se distingue des éléments du système contact par l’incapacité d’acquérir une activité du simple fait de son immobilisation sur une surface étrangère (contrairement au facteur XII, qui interagit avec de telles surfaces de manière directe – il en va de même pour la PK et le KHPM) (15). En revanche, le facteur XI peut être activé par sa propre forme activée (auto-activation) (12), propriété partagée avec le facteur XII et la PK.
Les environnements dans lesquels le facteur XI est activé, que ce soit par le XIIa ou par la thrombine, ne peuvent être que restreints, comme physiologiquement pour toutes les réactions de coagulation (16), mais ils restent encore mal définis.
La structure homodimèrique du facteur XI, caractéristique unique parmi tous les facteurs de la coagulation, favorise son activation (17). Elle serait aussi nécessaire à sa sécrétion, et permettrait, grâce au positionnement du site catalytique de chaque monomère, le clivage simultané en deux points du facteur IX, l’activant efficacement (17). De même, cela permettrait à une molécule de facteur XI de se lier à deux récepteurs cellulaires différents (18).
L’inhibiteur physiologique principal du facteur XIa, et aussi du facteur XIIa et de la kallicréine), n’est pas l’antithrombine, cette serpine ne contribuant que pour 1/6e de l’inhibition du facteur XIa – mais le C1-inhibiteur ; l’α1-antitrypsine (anti-protéinase à large spectre) joue aussi un rôle, ainsi que l’α2-antiplasmine, le ZPI (19) et la protéase nexine-II (20) ; et enfin l’inhibiteur versatile qu’est l’α2-macroglobuline est aussi impliqué dans l’ensemble de ces réactions, mais dans une bien moindre mesure (21,22).
RÔLE PHYSIOLOGIQUE DANS LA GÉNÉRATION DE THROMBINE
Nous avons vu pourquoi le facteur XI joue un rôle dans la coagulation débutant avec l’interaction du sang avec le FT, contrairement au système KKS ; mais comment intervient-il ?
Les raisons pour lesquelles le facteur XI est parfois, chez certains sujets et dans certaines circonstances, utile à une bonne hémostase, ne sont pas bien élucidées. Fait capital, cette contribution n’est pas prévisible par des explorations menées au laboratoire clinique, ce qui complique la gestion du risque hémorragique des déficits constitutionnels et aussi de celui induit par les traitements anticoagulants ciblant le facteur XI(a).
Deux formes de contribution ont été mises en avant. En premier lieu, la formation accrue de thrombine (surtout si le FT est en faible quantité) permise par la boucle de rétrocontrôle positif par la thrombine (thrombin begets thrombin) favorise l’activation du TAFI (inhibiteur de la
fibrinolyse activable par la trombine), retardant ainsi la fibrinolyse (23,24) ; cela contribue à expliquer les localisations préférentielles des saignements (au sein des tissus riches en activateurs du plasminogène) et suggère un intérêt clinique des tests explorant concomitamment la
coagulation et la fibrinolyse. Deuxièmement, le facteur XIa permet la propagation dans l’espace, par le biais d’expériences avec le dispositif ingénieux ThromboDynamics, où la coagulation démarre au contact de FT immobilisé, mimant ainsi, au moins en partie, la physiologie (25,26).
RÔLE IMPORTANT VOIRE DÉCISIF DES POLYPHOSPHATES
Les polyphosphates sont constitués de la répétition du motif phosphate, chargé négativement. La longueur des chaînes (linéaires) dépend de la source, plaquettaire ou exogène, bactérienne (27).
Les polyphosphates plaquettaires potentialisent des réactions de la phase contact et celles impliquant le facteur XI. S’ils sont de plus longue taille, ils sont supposés permettre l’activation du FXII (28,29), et même une
génération de thrombine débutée avec le facteur XIIa indépendamment du facteur XI (30), ce qui constituerait une limite aux approches anticoagulantes ciblant ce facteur.
INTERACTIONS AVEC DES CELLULES
Le facteur XI se lie à la GPIb plaquettaire (31). Une anomalie de l’activité procoagulante des plaquettes a d’ailleurs été notée très tôt en cas de maladie de Bernard-Soulier dans sa forme bi-allélique (32). Le facteur XI est présent dans les granules α (33), avec une quantité certes très minoritaire par rapport à celle dans le plasma, mais avec une situation privilégiée pour jouer un rôle clé dans la génération de thrombine, étant sécrété après activation plaquettaire (notamment par la thrombine qui interagit aussi avec GPIb). Ces interactions complexes (34) ont suscité des travaux qui n’ont pas toujours pu être confirmés, voire ont été rétractés (35). Le rôle des plaquettes dans l’activation du facteur XI reste donc une question imparfaitement résolue. Le CD36 plaquettaire, exprimé de manière constitutive à la surface des plaquettes, a été rapporté comme amplifiant la génération de thrombine dépendante du FXI lorsque les quantités de phospholipides procoagulants exposés sont limitées (36).
Une étude récente a étudié la liaison de facteur XIa aux plaquettes (qui n’implique pas la GPIb, contrairement à celle du facteur XI), et ses conséquences sur son activité : une protection vis-à-vis des effets inhibiteurs du sécrétome plaquettaire a pu être mise en évidence (34).
Le facteur XI(a) pourrait aussi interagir avec les cellules endothéliales (37) via le KHPM, comme pour la prékalli-créine. Pour cette dernière, il en résulterait son activation, ce qui pourrait constituer une voie physiologique d’activation par le système contact (14).
FACTEUR XI ET COAGULATION AU CONTACT DE SURFACES « ÉTRANGÈRES » : UN PROBLÈME PERSISTANT
Lorsqu’un dispositif médical vient au contact du sang, l’hémocompatibilité du matériel étranger – exogène – n’est jamais totale ; il y a donc activation de la coagulation, que ce soit en situation extracorporelle (risque de dépôts de fibrine compromettant le bon fonctionnement du dispositif –dialyseur, oxygénateur…) ou intracorporelle (risque de thrombose) (10). Pour dissocier complètement les effets anti-hémostatique et antithrombotique, le facteur XII(a) est alors la meilleure cible d’un inhibiteur thérapeutique, d’autant qu’une faible partie du facteur XIa formé semble, dans ces circonstances, suffisante pour une génération de thrombine efficace.
Cette activation inéluctable au contact d’une surface étrangère compromet l’étude fine de la coagulation en laboratoire. En fait le recueil du sang dans un tube contenant comme anticoagulant du citrate, son passage dans l’aiguille ou pire dans un raccord type « butterfly » (papillon), conduit inévitablement à l’activation de la phase contact, avec formation du XIa. Cela complique considérablement l’étude de la coagulation démarrée avec de très faibles concentrations de FT, celle de biomarqueurs d’activation in vivo (complexes enzyme – serpine tels que XIa-C1INH (38), pouvant être formés au cours de la phase pré-analytique et non pas in vivo), et l’étude fonctionnelle des vésicules extracellulaires exprimant du FT (39). L’activationnon souhaitée de la phase contact doit donc être freinée le plus vite possible (par addition de CTI). Qu’en est-il dans le format TC + A, pour explorer la coagulation par la voie intrinsèque ? Le TC + A étudie fonctionnellement le facteur XI, sa capacité à être activé par le facteur XIIa, et son action activatrice en cascade sur le facteur IX. La pré-incubation à 37 °C accentue délibérément ce phénomène ; lors de la recalcification, c’est la quantité de facteur XIa déjà formée qui détermine la force de l’enclenchement de la cascade avec ses étapes calcium-dépendantes (40). À noter que des variants du facteur XI dont l’activation par la thrombine serait défectueuse ne sont pas détectés au laboratoire en pratique courante.
L’implication du facteur XI dans la coagulation en condition de rétrocontrôle positif par la thrombine n’est pas explorée par le TC + A. Avec les tests dit de « génération de thrombine », il convient d’utiliser le FT à très faibles concentrations, et au mieux en présence de plaquettes (car il y a plusieurs interactions entre facteur XI et plaquettes qui influencent la génération de thrombine), ce qui complique fortement la démarche (10).
AUTRES INTERACTIONS ET EFFETS DU FACTEUR XIa
Le facteur XIa n’est pas « monogame » et a donc plusieurs « partenaires » − il existe une promiscuité enzymatique (7) :
il agit sur d’autres protéines, comme dans le système hémostatique les facteurs V et VIII qu’il active (41) ; avec ADAMTS13 qu’il clive des domaines en position C-terminale, ce qui rend ADAMTS13 moins active (42) ; enfin, il inhibe le TFPI.
Il interagit aussi avec le complément avec des impacts sur les processus inflammatoires. Par exemple il a été rapporté que le facteur XIa agit sur le système du complément, en inhibant le factor H (CFH), ce qui amplifie la voie alterne (43) en se liant à un autre régulateur, la properdine (44).
L’activation du facteur XIa par la thrombine peut être inhibée par la β2-glycoprotéine I (β2GPI), qui en retour peut être clivée à son extrémité C-terminale avec une altération de ses propriétés anticoagulantes (45,46). Une autre protéine de l’inflammation est la cible du facteur XIa : la prochemerin ; sa forme activée (chemoattractant and adipokine chem158K) contribue au recrutement des leucocytes dans le thrombus, à la prolifération des cellules musculaires lisses et des adipocytes et à l’apoptose des cellules endothéliales (47).
EFFETS VASCULAIRES
Il a été rapporté que le facteur XIa exerce aussi des effets (directs et indirects) sur la paroi vasculaire (48). À titre d’exemples : 1) il accroît la perméabilité de l’endothélium en clivant le domaine extracellulaire de la VE-cadhérine. Il s’agit d’un phénomène essentiel au développement de l’inflammation (49) ; 2) Il est capable de se fixer via son domaine A2 à la laminine, composant important de la matrice extracellulaire ; 3) Il peut agir directement sur les cellules musculaires lisses des vaisseaux via le récepteur PAR-1, provoquant une entrée de calcium et une accélération de la migration cellulaire en cas de blessure vasculaire (50).
Le facteur XIa interagit avec le PAI-1 endothélial, ce qui l’inhibe (51). Le phénomène est complexe car à l’inverse l’endothélium favorise l’activation du système KKS.
Un autre exemple illustre le pléiotropisme du facteur XI : il pourrait jouer un rôle protecteur dans l’insuffisance cardiaque à fonction systolique conservée (52). L’impact clinique de l’inhibition du facteur XI peut être, en conséquence, jugé préoccupant à cet égard, et une association entre niveau bas de facteur XI, insuffisance cardiaque, et anomalies de l’oreillette gauche a été démontrée (53).
Il découle de l’existence de toutes ces interactions, fermement démontrées ou à confirmer, qu’inhiber le facteur XI(a) est susceptible de modifier divers équilibres subtils, au-delà de la génération de thrombine… Il n’est notamment pas encore établi que l’inflammation est réduite après administration de tels médicaments. Et en ce sens l’exploration au laboratoire restreinte au TC + A, et aux mesures du facteur XI:C, de l’activité anti-XIa… passe complètement à côté de ces éventuels effets de l’inhibition du facteur XI(a).
FACTEUR XI ET THROMBOSES
Les arguments classiques en faveur de l’implication du facteur XI dans les thromboses sont expérimentaux (modèles animaux) et épidémiologiques : un facteur XI bas est protecteur (54), tandis qu’un facteur XI élevé est associé à un risque accru de thrombose (55-57).
Des accidents thrombotiques veineux et même artériels ont été observés chez des patients déficitaires en facteur XI (souvent avec facteurs de risque de thrombose) traités avec un concentré de facteur XI plasmatique pouvant conduire à des niveaux trop élevés de facteur XI et de biomarqueurs d’activation de la coagulation (58). Des niveaux élevés de facteur XI activé sont en effet associés à des phénomènes prothrombotiques (59,60). Le rôle thrombogène du FXI, dans sa forme activée (FXIa), est conforté par la survenue de thromboses dans les 24 h suivant la perfusion d’IgG polyvalentes, des traces de FXIa dans ces concentrés ayant été impliquées dans la survenue de ces complications (61). Les recommandations actuelles requièrent la mesure du FXIa dans ces concentrés, et s’il est présent au-dessus d’un certain seuil, les lots doivent être rejetés.
Sur le plan mécanistique, durant la phase aiguë de la thrombose veineuse, le facteur XI intervient en association avec diverses protéines impliquées dans la réponse inflammatoire (thrombo-inflammation), interagissant avec la matrice extracellulaire, le métabolisme lipidique ou l’apoptose (62). L’implication du facteur XI dans les thromboses débutant dans un foyer thrombogène contenant peu de FT serait déterminée par la formation de NETs et par la présence de polyphosphates de très grande longueur (d’origine bactérienne notamment), qui sont capables d’activer le facteur XII, celui-ci pouvant même déclencher la génération de thrombine sans le concours du facteur XI (30), autre limite potentielle du ciblage de ce dernier facteur pour combattre la thrombose.
En situation physiologique, seuls les polyphosphates issus des granules denses plaquettaires sont présents au site de la brèche vasculaire, du fait de l’activation et de la sécrétion plaquettaires ; comme mentionné plus haut, leur taille réduite expliquent qu’ils sont seulement capables de faciliter l’activation du facteur XI mais non celle du facteur XII.
En situation prothrombotique, lorsque le FT est présent en grandes quantités, le rôle du facteur XI pourrait n’être qu’accessoire ; si les NETs et les polyphosphates de très grande taille interviennent, le facteur XI(a) joue alors un rôle déterminant en aval du facteur XII(a), comme au cours du sepsis, et éventuellement d’autres contextes inflammatoires.
UN PEU DE PHARMACOLOGIE : HÉPARINE ET APROTININE
Fait capital, l’héparine n’est pas un bon inhibiteur du facteur XI(a) : bien qu’elle ait été identifiée comme pouvant accélérer l’inactivation de sa forme activée par l’antithrombine (63),
en système purifié surtout (64), elle est en fait incapable d’accélérer l’effet de l’antithrombine sur la forme activée (65), mais elle pourrait même favoriser son activation – par la thrombine liée à la fibrine (66).
L’aprotinine est utilisée comme antifibrinolytique, inhibiteur de la plasmine, mais a de nombreuses autres cibles, dont le facteur XIa (67).
Les médicaments anticoagulants ciblant le facteur XI(a) sont abordés dans un autre article de ce dossier thématique.
POINTS CLÉS À RETENIR
• Le facteur XI, activé par le XIIa, articule le système contact avec la ténase intrinsèque (voie intrinsèque de la coagulation).
• Le facteur XI est aussi activé par la thrombine (une des boucles de rétrocontrôle positif de la génération de thrombine), et il joue de ce fait un rôle dans l’hémostase physiologique (la protection contre les saignements, et leur arrêt) démarrée par le facteur tissulaire (FT), certes limité mais parfois utile voire essentiel.
• Le facteur XIa active le facteur IX, propriété qu’il partage avec le facteur VIIa (en présence de FT – boucle de Josso).
• Le facteur XIa agit directement ou indirectement sur d’autres protéines que le facteur IX, comme les facteurs V et VIII (activation), le TFPI (inhibition), et ADAMTS13 (effet complexe). Il a aussi des effets sur la paroi vasculaire.
• L’inhibiteur physiologique principal du facteur XIa n’est pas l’antithrombine, mais le C1-inhibiteur.
• Les raisons pour lesquelles le facteur XI peut être utile à une bonne hémostase ne sont pas totalement élucidées (retardation de la fibrinolyse, extension dans l’espace…). Cette implication n’est pas prévisible par des explorations au laboratoire. Le facteur IX peut être activé par la kallicréine, court-circuitant ainsi les facteurs XII et XI. Cette voie courte pourrait expliquer en partie la variabilité du phénotype clinique (hémorragique) des déficits constitutionnels en facteur XI.
• L’implication du facteur XI dans les thromboses semble dépendre de la disponibilité de polyphosphates et de leur longueur de chaîne, et de la formation de NETs.
• L’activation délétère de la coagulation au contact de surfaces « étrangères » (dispositifs médicaux placés au contact du sang) implique les facteurs XII et XI.
• L’activation du système contact et du facteur XI dès le prélèvement de sang constitue un véritable poison pour l’étude fine de la coagulation.
• Le TC + A étudie fonctionnellement le facteur XI, plus précisément sa capacité à être activé par le facteur XIIa, et son action activatrice en cascade sur le facteur IX.
• L’exploration au laboratoire de l’implication du facteur XI dans la coagulation en condition de rétrocontrôle positif par la thrombine doit se faire en utilisant de très faibles concentrations de FT, et au mieux en présence de plaquettes.