RÉSUMÉ
L’hémophilie A acquise (AHA) est une pathologie auto-immune rare, dont l’incidence est estimée entre 1 et 1,5 cas par million d’habitants. Le diagnostic peut être posé de manière fortuite, mais il est le plus souvent motivé par la survenue de saignements inhabituels. Dans seulement 50 % des cas, une étiologie sous-jacente est identifiée, incluant notamment des maladies auto-immunes, des néoplasies ou la grossesse. L’évolution de l’AHA est généralement brutale et sévère, en particulier chez les patients âgés et polymorbides, avec un taux de mortalité avoisinant les 15 %. Ce risque est principalement imputable à la pathologie causale, mais également aux effets délétères de l’immunosuppression. Nous rapportons ici un cas d’AHA survenu dans le contexte d’une pancréatite auto-immune de type 2 nouvellement diagnostiquée, et proposons une revue de la littérature des cas associant AHA et pancréatite auto-immune.MOTS CLÉS
hémophilie A acquise, pancréatite auto-immune
ABSTRACT
Acquired hemophilia A (AHA) is a rare autoimmune disease, with an estimated incidence of 1 to 1.5 cases per million inhabitants. The diagnosis may be made incidentally, but it is most often prompted by the occurrence of unusual bleeding. An underlying etiology is identified in only 50% of cases, most commonly autoimmune diseases, malignancies, or pregnancy. The course of AHA is typically sudden and severe, particularly in elderly patients with multiple comorbidities, with a mortality rate approaching 15%. This risk is mainly attributable to the underlying condition, but also to the adverse effects of immunosuppressive therapy. We report here a case of AHA occurring in the context of newly diagnosed type 2 autoimmune pancreatitis and provide a review of the literature on cases associating AHA with autoimmune pancreatitis.
KEYWORDS
acquired hemophilia A, autoimmune pancreatitis
INTRODUCTION
L’hémophilie A acquise (AHA) est une maladie autoimmune rare, caractérisée par le développement d’autoanticorps circulants inhibant le facteur VIII (FVIII). Son incidence est estimée entre 1 et 1,5 cas par million d’habitants, avec une prévalence plus élevée chez les patients âgés (1).Dans la majorité des cas, l’AHA est suspectée face à l’apparition soudaine de manifestations hémorragiques sévères et spontanées, principalement chez des personnes âgées sans antécédents personnels ou familiaux de troubles de la coagulation. Dans certains cas, le diagnostic est posé en l’absence de symptômes hémorragiques évidents et est suggéré par un allongement marqué du temps de thromboplastine partielle activée (aPTT) (2).
Les manifestations cliniques de l’AHA sont dominées par les hématomes musculaires et des tissus mous, souvent volumineux, pouvant entraîner une anémie sévère et/ou une compression nerveuse. Contrairement à l’hémophilie A congénitale (HA), les hémarthroses sont rares dans l’AHA. D’autres symptômes peuvent inclure des saignements muqueux (épistaxis, gingivorragies, métrorragies) ainsi que des hémorragies sévères engageant le pronostic vital, telles que les hémorragies gastro-intestinales, les hématomes rétropéritonéaux et les hémorragies intracrâniennes (3).
Environ la moitié des cas d’AHA sont associés à une pathologie sous-jacente, notamment des maladies auto-immunes, des cancers, la prise de certains médicaments, la grossesse ou le post-partum. Dans l’autre moitié des cas, aucune cause sous-jacente n’est identifiée (4). L’identification des patients atteints d’AHA est complexe en raison de la diversité des présentations cliniques et du fait que cette affection reste méconnue des médecins généralistes et spécialistes, ce qui entraîne fréquemment des retards diagnostiques et thérapeutiques. L’AHA est associée à une mortalité élevée, en particulier chez les patients âgés. Si les hémorragies sont responsables de moins de 10 % des décès, elles entraînent une morbidité importante. La principale cause de décès est souvent liée aux complications du traitement immunosuppresseur ou à la pathologie sous-jacente (5).
La pancréatite auto-immune (PAI) est une forme rare d’inflammation pancréatique, qui peut se manifester sous la forme d’une masse focale ou d’une infiltration diffuse. Le type 1, le plus fréquent, s’inscrit dans le cadre des maladies liées aux IgG4, un groupe d’affections fibro-inflammatoires systémiques pouvant atteindre plusieurs organes. Le type 2,plus rare, est associé dans 20 à 30 % des cas à une maladie inflammatoire chronique de l’intestin (6).
L’association entre AHA et PAI a rarement été rapportée dans la littérature médicale. Nous décrivons ici un cas d’AHA survenant dans un contexte de PAI de type 2, sans atteinte d’autre organe.
DESCRIPTION DU CAS
Il s’agit d’un patient de 63 ans, récemment diagnostiqué d’une pseudo-tumeur pancréatique, initialement suspectée d’être un adénocarcinome. Il a bénéficié d’une duodéno-pancréatectomie céphalique en juin 2022. L’examen anatomopathologique a mis en évidence une pancréatite chronique associée à une fibrose intense centrée sur les canaux, avec des infiltrats granulocytaires intra-épithéliaux étendus, sans augmentation significative des plasmocytesIgG4, évoquant fortement une PAI de type 2.
Un mois après l’intervention, le patient est hospitalisé en urgence pour une hémorragie digestive haute sur ulcère gastrique anastomotique Forrest 1B, dans un contexte de choc hémorragique. Il est traité par injection d’adrénaline etpose de clips hémostatiques. Au total, il reçoit 10 concentrés érythrocytaires, 1 plasma frais congelé et 2 g d’Haemocomplettan®.
Un allongement de l’aPTT à 81 secondes (norme :26-37 secondes) est constaté, alors qu’il était court un mois plus tôt (24,4 secondes). Le TP (Quick) était normal à 95 % (norme : >70 %), et le fibrinogène à 3,8 g/L(norme : 2,0-4,0 g/L). Le facteur VIII était abaissé à 3 %,tandis que le facteur von Willebrand était supérieur à200 %. Les facteurs IX, XI et XII étaient également dans les normes. Enfin, la présence d’un inhibiteur du facteur VIIIà 1,4 UB a permis de confirmer le diagnostic d’une AHA. L’hémogramme à l’admission mettait en évidence une anémie avec 65 g/L d’hémoglobine, sans autres cytopénies. Un traitement par FVIII porcin recombinant (Obizur®) à100 UI/kg est initié en urgence, permettant une excellente réponse biologique initiale. Le traitement est poursuivi à 50 UI/kg deux fois par jour pendant quatre jours, mais l’efficacité devient transitoire malgré une augmentation de la posologie à 100 UI/kg, suggérant l’apparition d’un anticorps inhibiteur croisé (non recherché). Au total, le patient reçoit 55 250 UI d’Obizur® durant une semaine.
En parallèle, une corticothérapie par prednisone à raison de 1 mg/kg a été instaurée dès l’admission, entraînant une amélioration progressive du taux de facteur VIII, qui a atteint 40 % au bout de 7 jours, permettant l’arrêt de la substitution. De plus, une réduction du titre d’inhibiteur a été observée, avec un taux de 0,55 UB à 3 semaines et indétectable à 6 semaines de traitement. La posologie a été maintenue pendant 4 semaines, avec un traitement bien toléré, à l’exception d’une décompensation diabétique survenue dans un contexte de diabète lié à une insuffisance pancréatique et du traitement de prednisone.
Sur le plan étiologique, une infection asymptomatique par le SARS-CoV-2 a été évoquée mais jugée peu probable. L’origine auto-immune semblait la plus plausible, dans le contexte d’une PAI récemment diagnostiquée. Le bilan immunologique révèlera une légère hypogammaglobulinémie (IgG à 6,2 g/L) avec le sous-type IgG4 dans la norme. L’électrophorèse des protéines ne montrait pas de monoclonalité. Aucun signe clinique ou biologique n’évoquait une autre maladie auto-immune sous-jacente. Le scanner thoraco-abdominal ne retrouvait pas de néoplasie. Le bilan infectieux (hépatites B et C, HSV, VZV) était négatif. La cytométrie en flux ne révèlera aucune population lymphocytaire monoclonale. Une rectosigmoïdoscopie est réalisée sans argument pour une maladie inflammatoire chronique intestinale. Aucun nouveau traitement potentiellement inducteur d’AHA n’a été introduit en post-opératoire. Le patient a poursuivi un sevrage progressif de la corticothérapie, aboutissant à un arrêt complet au bout de 4 mois. Le taux de facteur VIII s’est maintenu au-dessus de 150 %, et l’inhibiteur a disparu, témoignant d’une réponse complète. Par ailleurs, le suivi gastro-entérologique s’est révélé tout à fait satisfaisant, sans récidive locale, ni signe de rechute de l’AHA à 3 ans de la présentation.
DISCUSSION
Nous rapportons ici un cas exceptionnel d’hémophilie A acquise (AHA), le plus probablement secondaire à une PAI. Bien que des cas similaires aient déjà été décrits dans la littérature chez l’adulte (Tableau 1), notre observation se distingue par le fait qu’il s’agit, à notre connaissance, du seul patient présentant une PAI de type 2.
Tableau 1 : Cas rapportés d’hémophilie A acquise associée à une pancréatite auto-immune dans la littérature.
Table 1: Reported cases of acquired hemophilia A associated with autoimmune pancreatitis in the literature.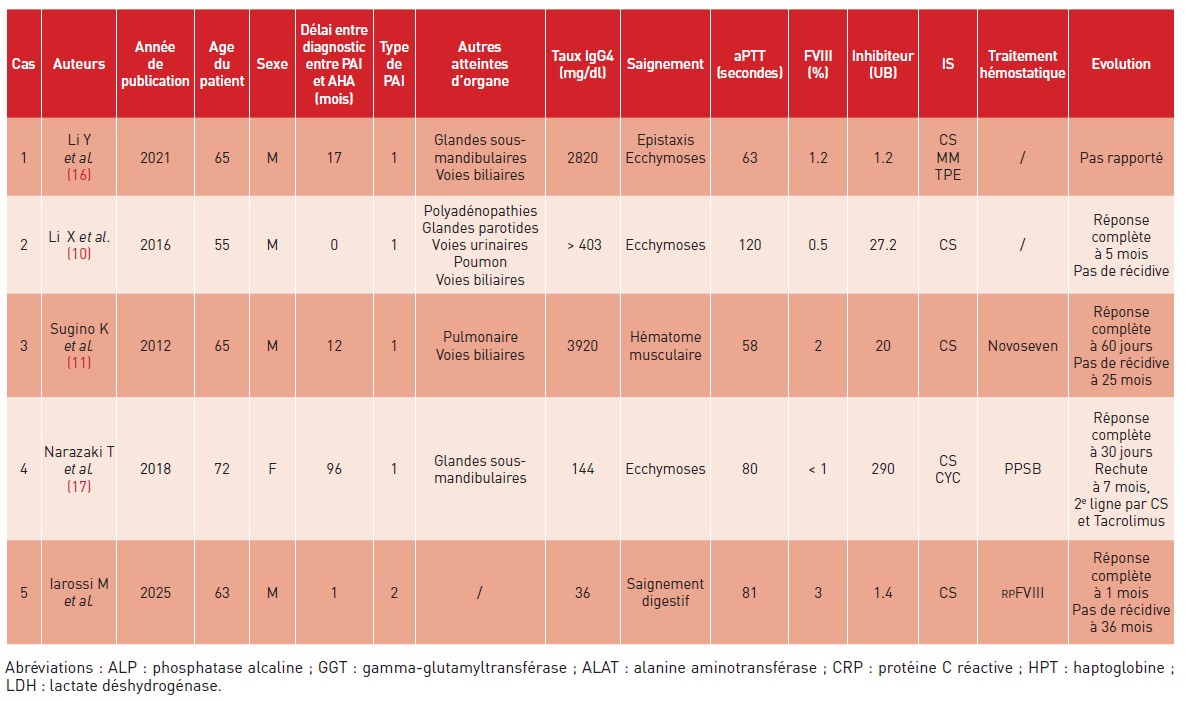
Dans l’AHA, le développement spontané d’auto-anticorps dirigés contre le FVIII neutralise son activité. Bien que la présence d’anticorps se liant au FVIII soit rapportée chez environ 19 % des individus en bonne santé, ces anticorps ciblent des épitopes non fonctionnels du FVIII et n’ont pas de pouvoir neutralisant. Chez les sujets sains, les principales sous-classes d’IgG dirigées contre le FVIII sont les IgG1 et IgG3, généralement à faibles titres. En revanche, chez les patients atteints d’AHA, les anticorps appartiennent principalement aux sous-classes IgG1 et IgG4, avec des titres bien corrélés à ceux des inhibiteurs (7).
Les PAI de type 1 sont décrites depuis 1993 et s’inscrivent dans le cadre des maladies liées aux IgG4. Elles sont caractérisées par une infiltration lymphoplasmocytaire dense comportant des plasmocytes IgG4-positifs, une fibrose, et répondent à des critères diagnostiques précis : tuméfaction diffuse ou localisée, masses touchant un ou plusieurs organes à l’examen clinique, taux sérique élevé d’IgG4 (≥ 135 mg/dl), infiltration marquée de lymphocytes et de plasmocytes, fibrose, ainsi que la présence de plasmocytes IgG4-positifs avec un ratio IgG4/IgG > 40 % et plus de 10 plasmocytes IgG4-positifs par champ à fort grossissement (HPF) à l’histologie (8). La PAI de type 2 touche des patients significativement plus jeunes, généralement dans la 3e ou 4e décennie de vie, avec une répartition équilibrée entre les sexes. Aucun critère de classification n’a été établi pour ce type, et aucun biomarqueur spécifique n’est actuellement disponible. Le diagnostic repose donc sur l’histologie, qui révèle un processus fibro-inflammatoire affectant principalement les canaux pancréatiques, avec une infiltration neutrophile des canaux de taille moyenne et petite. Le trait histologique le plus distinctif est la présence de lésions épithéliales granulocytaires dans ces canaux, et parfois au niveau des acini. Contrairement à la PAI de type 1, les plasmocytes IgG4-positifs sont absents ou rares. De plus, contrairement au type 1 qui s’associe souvent à des atteintes multi-organes, le type 2 est généralement isolé, bien qu’environ 15 % des cas présentent une maladie inflammatoire chronique de l’intestin concomitante (9).
Il est essentiel de distinguer les sous-types de PAI, car bien que l’approche clinique et la stratégie thérapeutique initiale restent similaires, la réponse au traitement est presque toujours favorable dans le type 2. Une absence de réponse efficace doit faire suspecter un autre diagnostic. Par ailleurs, dans la PAI de type 2, les rechutes ne sont pas fréquentes (9).
Sanges et al. ont étudié la relation immunopathogénique entre l’AHA et la PAI, en explorant deux hypothèses physiopathologiques : d’une part, une perte de tolérance envers le FVIII endogène, induite par une prolifération oligoclonale de plasmocytes IgG4+, aboutissant à la production d’auto-anticorps anti-FVIII ; d’autre part, une prolifération polyclonale de plasmocytes IgG4+, responsable d’une augmentation de l’activité anti-FVIII collatérale, normalement présente à des niveaux indétectables. Aucun de ces mécanismes n’a toutefois pu être formellement confirmé (7). Notre observation d’une PAI de type 2, sans élévation des IgG4, suggère que, bien qu’un rôle pathogénique des IgG4 puisse être impliqué dans certains cas, d’autres mécanismes physiopathologiques doivent également être envisagés dans l’apparition de l’AHA dans le contexte d’une PAI.
Le traitement de l’AHA nécessite une approche multidisciplinaire, reposant sur plusieurs piliers essentiels : éviter les gestes à risque hémorragique, contrôler les épisodes de saignement aigu, éradiquer les inhibiteurs afin de prévenir les récidives, et prendre en charge la pathologie sous-jacente.
La gestion concomitante de l’AHA et de la PAI repose sur l’immunosuppression, avec pour objectif l’éradication des auto-anticorps. Le schéma thérapeutique classique repose sur l’utilisation de corticostéroïdes, de cyclophosphamide ou de rituximab, seuls ou en association (4). Conformément aux recommandations, nous avons opté pour une première ligne de traitement par corticostéroïdes seuls, compte tenu du taux initial de FVIII et du titre d’inhibiteur. Cette stratégie a permis d’obtenir une réponse rapide et une rémission complète, avec normalisation des taux de FVIII et absence de récidive à distance. Elle s’est également révélée efficace dans deux autres cas rapportés (10,11).
Plusieurs agents peuvent être utilisés pour maîtriser les saignements dans l’AHA, notamment les agents bypassants tels que le concentré de complexe prothrombique activé (aPCC) et le facteur VII activé recombinant (rFVIIa), ainsi que le facteur VIII humain (en présence d’un taux d’inhibiteur faible) ou porcin. Il n’existe pas de préférence clairement établie entre ces produits, le choix dépendant essentiellement de leur accessibilité et des habitudes du centre et du taux d’inhibiteur. Bien que ces traitements soient efficaces pour restaurer l’hémostase, ils présentent certaines limites : administration exclusivement intraveineuse, demi-vie courte nécessitant des injections répétées, efficacité hémostatique parfois incomplète, surveillance biologique complexe, risque thrombogène non négligeable (aPCC et rFVIIa), et contraintes en cas de traitement au long cours (12).
L’émergence de nouveaux agents thérapeutiques, tels que l’émicizumab, a profondément modifié la prise en charge de l’HA, au point d’en devenir le traitement de référence chez les patients atteints de formes sévères, avec ou sans inhibiteurs (13). Bien que ce traitement ne fasse pas encore partie des recommandations officielles dans l’AHA, plusieurs cas cliniques ont rapporté son efficacité dans cette indication, et son utilisation est actuellement en cours d’évaluation dans plusieurs études prospectives (14), afin de confirmer les observations décrites par Shima et al. (15). L’émicizumab, en tant qu’agent prophylactique, pourrait permettre une approche thérapeutique moins agressive de l’éradication des inhibiteurs, limitant ainsi l’exposition aux immunosuppresseurs et les effets indésirables qui leur sont associés (14).
CONCLUSION
Nous rapportons un cas unique d’AHA survenue dans le contexte d’une PAI de type 2, ce qui contraste avec les rares cas précédemment décrits dans la littérature, où l’AHA était associée à une PAI de type 1 et une maladie à IgG4. Le traitement par corticostéroïdes seuls a permis d’obtenir une rémission complète, sans récidive à distance, comme rapporté dans deux autres observations cliniques similaires.
POINTS CLÉS À RETENIR
• Premier cas rapporté d’AHA survenant dans le contexte d’une PAI de type 2, avec une absence d’élévation du taux IgG4, suggérant un mécanisme physiopathologique distinct.
• Approche multidisciplinaire nécessaire qui repose sur l’éradication des inhibiteurs, le contrôle des saignements et le traitement de la pathologie sous-jacente, nécessitant une coordination étroite entre spécialités.
• L’émicizumab émerge comme une option prometteuse dans l’AHA, bien que hors recommandations actuelles.
Liens d’intérêts : cet article est une traduction de l’article original publié dans Hämostaseologie, intitulé « An unusual association between acquired Hemophilia A and type 2 autoimmune pancreatitis » (DOI : 10.1055/a-2829-7111).
Alessandro CASINI déclare avoir des liens d’intérêts avec LFB, Novo Nordisk et Sobi ; Pierre FONTANA déclare avoir des liens d’intérêts avec Novo Nordisk, Roche, Sobi et Takeda. Les autres auteurs déclarent ne pas avoir d’autre lien d’intérêt en rapport avec cet article.