RÉSUMÉ
La Revue Francophone d’Hémostase et Thrombose vous propose, outre la réception trimestrielle de la revue, des publications online régulières sur la plateforme www.rfht.fr sous la forme : • d’une veille bibliographique avec les meilleurs articles publiés dans nos spécialités ; • d’actualités commentées offrant une vision critique d’une sélection d’articles internationaux et leur mise en perspective en pratique clinique. Vous trouverez ci-après une sélection de ces articles récents, décryptés, analysés et commentés pour vous par Antoine Babuty, Camil Chiali, Laure Macraigne et Geneviève McCluskey.En partenariat avec ![]()

La Revue Francophone d’Hémostase et Thrombose vous propose, outre la réception trimestrielle de la revue, des publications online régulières sur la plateforme www.rfht.fr sous la forme :
• d’une veille bibliographique avec les meilleurs articles publiés dans nos spécialités ;
• d’actualités commentées offrant une vision critique d’une sélection d’articles internationaux et leur mise en perspective en pratique clinique.
Vous trouverez ci-après une sélection de ces articles récents, décryptés, analysés et commentés pour vous par Antoine Babuty, Camil Chiali, Laure Macraigne et Geneviève McCluskey.
UN NANOBODY BISPÉCIFIQUE POUR LE TRAITEMENT DE LA MALADIE DE WILLEBRAND DE TYPE 1
D’après : A bispecific nanobody for the treatment of von Willebrand disease type 1. Peyron I, Casari C, McCluskey G, Roullet S, Licari VN, Bocquet E, et al. Blood 2025 [Online ahead of print].
Actualité commentée réalisée par Antoine BABUTY
Justificatifs et objectifs
La maladie de Willebrand (VWD) de type 1 est un trouble de l’hémostase caractérisé par un déficit quantitatif partiel en facteur von Willebrand (VWF), entraînant des symptômes hémorragiques et une altération de la qualité de vie. Les traitements actuels comprennent principalement l’utilisation de la desmopressine ou les concentrés de VWF. Les auteurs ont développé un nanobody bispécifique, le KB-V13A12, qui lie le VWF et l’albumine, permettant le recyclage du VWF par le récepteur Fc néonatal (FcRn) et ainsi augmente sa demi-vie (1). L’objectif de cette étude était d’évaluer si le KB-V13A12 pouvait augmenter les taux de VWF circulant et améliorer l’hémostase.
Méthodes
Le KB-V13A12 a été injecté par voie intraveineuse ou sous-cutanée à des souris déficientes en VWF humanisé (souris hVWD-1). Les taux résiduels plasmatiques de KB-V13A12 étaient déterminés par western-blot. La clairance du VWF était évaluée après l’administration de VWF recombinant et de KB-V13A12 chez les souris hVWD-1, via le dosage de l’activité (VWF:Act), de l’antigène (VWF:Ag), des multimères, du propeptide (VWF:pp) du VWF ainsi que le dosage du facteur VIII (FVIII). La fonction hémostatique était évaluée par ponction de la veine saphène et par des tests de perfusion sur une matrice de collagène fibrillaire dans des conditions de flux artériel.
Résultats
Tout d’abord, les auteurs ont démontré que la liaison du nanobody aux VWF et à l’albumine n’affecte pas la liaison du VWF à la GpIBα plaquettaire ou au FVIII. Quatorze jours après l’injection du nanobody (5 mg/ml), les taux de VWF:Ag étaient significativement augmentés (x1,4, p = 0,0026) ainsi que les taux de FVIII (x1,5) comparativement aux souris non traitées. Le ratio VWF:pp/VWF:Ag était significativement diminué après injection du KB-V13A12 suggérant une diminution de la clairance du VWF et donc un recyclage plus important. Ce résultat a été confirmé chez des souris déficientes en FcRn, puisque cette diminution de clairance n’a pas été retrouvée. Le traitement par KB-V13A12 permettait, après perfusion du sang sur une surface recouverte de collagène, une augmentation de l’adhérence plaquettaire
2 fois plus importante. Après ponction de la veine saphène, le nombre de caillots formés ainsi que le temps de saignement maximal se sont normalisés chez les souris hVWD-1 après injection de KB-V13A12 comparativement à des souris non traitées et des souris sauvages.
Avis d’expert
Cette étude est la deuxième décrivant l’allongement de la demi-vie du VWF via l’albumine (2). Le nanobody décrit dans cette étude se lie avec une forte affinité à l’albumine, ainsi il circule entièrement lié à celle-ci. L’albumine étant présente à forte concentration dans le plasma, ses variations de concentration n’affecteront pas la formation de complexe avec le nanobody. Ce nanobody se lie également avec une forte affinité au VWF, et il était estimé que des concentrations supérieures à 50 nM permettraient de lier plus de 90 % du VWF endogène.
Concernant la résistance du complexe albumine-nanobody-VWF à la dissociation induite par un pH faible dans les endosomes précoces, les tests in vitro montraient une association du VWF au reste du complexe à pH 5,6 ou 7,4. Cette propriété permet un recyclage efficace du VWF via le récepteur FcRn, mise en évidence via une diminution du ratio VWFpp/VWF:Ag, puisque la synthèse du propeptide reste inchangée, et par une diminution de la clairance du nanobody dans les souris FcRn-/- comparée aux souris sauvages. Les conditions de stockage du VWF dans les corps de Weibel-Palade se font à des pH plus acides que dans les endosomes précoces, rendant peu probables des altérations structurales du VWF (3), ce qui a été confirmé par l’absence de modification de l’étude des multimères du VWF lié au nanobody. Enfin, ce nanobody étant fortement humanisé, aucun anticorps anti-nanobody n’a été détecté dans une expérience de dosage répété suggérant un faible potentiel immunogène de la molécule.
Des études supplémentaires chez l’humain devront être réalisées afin de savoir si l’augmentation des taux de VWF sera suffisante pour avoir un impact sur le profil hémorragique. Il est important de souligner que ce traitement ne sera probablement pas approprié pour les déficits
quantitatifs ou qualitatifs importants en VWF. Ce traitement aurait toutefois l’énorme avantage d’avoir un faible impact sur la charge liée au traitement de la maladie via son mode d’administration sous-cutanée et sa demi-vie longue permettant des injections une ou deux fois par mois.
Par ailleurs, l’utilisation de cette stratégie pourrait être étendue à d’autres pathologies associées à une déficience quantitative en protéines plasmatiques circulantes.
Références
1. McCluskey G, Heestermans M, Peyron I, Pascal E, Clavel M, Bun E, et al. A fully humanized von Willebrand disease type 1 mouse model as unique platform to investigate novel therapeutic options. Haematologica 2025 ; 110 : 923-37.
2. Schulte S. Innovative coagulation factors: albumin fusion technology and recombinant single-chain factor VIII. Thromb Res 2013 ; 131 : S2-6.
3. Terglane J, Menche D, Gerke V. Acidification of endothelial Weibel-Palade bodies is mediated by the vacuolar-type H+-ATPase. PLoS One 2022 ; 29 : e0270299
FACTEURS PRONOSTIQUES DE RÉCIDIVE DANS L’HÉMOPHILIE A ACQUISE : RÉSULTATS D’UNE ÉTUDE OBSERVATIONNELLE à LONG TERME
D’après : Prognostic factors for recurrence in acquired hemophilia: A results from a long-term observational study.
Reich L, Gatzke F, Rauchfuss S, Roth S, Miesbach W. Res Pract Thromb Haemost 2025 ; 9 : 102707.
Actualité commentée réalisée par Camil CHIALI
Justificatifs et objectifs
L’hémophilie A acquise (HAA) est une maladie auto-immune rare due à des auto-anticorps dirigés contre le facteur VIII (FVIII), entraînant des saignements sévères.
Malgré l’efficacité des traitements immunosuppresseurs, environ 20 à 30 % des patients présentent une rechute.
Or, peu de données existent sur les facteurs prédictifs de récidive. L’objectif de cette étude est d’identifier les paramètres cliniques et biologiques associés à un risque accru de rechute après une première rémission, dans le but d’optimiser le suivi et la stratégie thérapeutique des
patients atteints d’HAA.
Méthodes
Cette étude observationnelle monocentrique a inclus 41 patients atteints d’HAA, définie par une activité du FVIII < 50 UI/dL et un inhibiteur ≥ 0,4 BU/mL. La rémission partielle (RP) se définit par l’absence de saignement après l’arrêt du traitement hémostatique (> 24 h) et un FVIII > 50 UI/dL. La rémission complète (RC) y ajoute une recherche d’inhibiteur négative, prednisolone < 15 mg/j et arrêt de toute autre immunosuppression. La rechute correspond à la réapparition des critères biologiques diagnostiques après RP ou RC. Une analyse univariée puis multivariée a été réalisée sur 11 variables cliniques et thérapeutiques.
Résultats
Parmi les 41 patients suivis, 18 (44 %) ont rechuté dans l’année suivant la rémission. Deux facteurs ont été identifiés comme significativement associés au risque de récidive : un taux initial de FVIII < 1 UI/dL (OR 0,32 ; IC 95 % [0,12-0,84]) et l’absence de rémission complète initiale (OR 0,30 ; IC 95 % [0,11-0,80]). Ces deux variables sont restées significatives en analyse multivariée : OR respectifs 0,37 [0,14-0,98] et 0,35 [0,13-0,93] (Tableau 1). Aucun des autres critères étudiés (titre de l’inhibiteur, âge, sexe, comorbidités, traitement initial, etc.) n’a montré de lien significatif avec la rechute.
Tableau 1 : Facteurs prédictifs de récidive – analyse multivariée.
Table1: Predictors of recurrence – multivariate analysis.

Avis d’expert
Cette étude présente un réel intérêt clinique en apportant des éléments concrets pour mieux anticiper les récidives d’HAA, une pathologie rare mais à fort potentiel morbide. L’identification de deux facteurs de risque simples à mesurer – le taux initial de FVIII et le statut de rémission complète – pourrait permettre un suivi plus ciblé. En pratique, un patient ayant un FVIII < 1 UI/dL ou n’ayant pas atteint une RC complète devrait faire l’objet d’un suivi rapproché pendant la première année, période durant laquelle toutes les rechutes ont été observées dans cette cohorte.
Cependant, il est important de noter certaines limites. L’échantillon est réduit, ce qui fragilise la puissance statistique. De plus, la sélection des patients (exclusion des décès précoces et des non-rémissions) introduit un biais favorable. Malgré cela, les résultats sont cohérents avec les données de la littérature antérieure. Par exemple, l’étude GTH-AH a déjà montré qu’un FVIII < 1 UI/dL était lié à un pronostic défavorable (1). Le suivi du ratio FVIII:C/VWF:Ag identifié par Trossaert et al. (2) comme potentiel facteur de risque de récidive, n’a pas été étudié ici.
En conclusion, même si cette étude n’apporte pas encore un modèle prédictif robuste, elle identifie deux facteurs de risque de récidive simples et utiles pour la pratique clinique dans l’HAA. L’arrivée de traitements comme l’emicizumab devrait néanmoins modifier le pronostic et la surveillance de cette pathologie.
Références
1. Tiede A, Klamroth R, Scharf RE, Trappe RU, Holstein K, Huth-Kühne A, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH-AH 01/2010 study. Blood 2015 ; 125 : 1091-7.
2. Trossaert M, Graveleau J, Thiercelin-Legrand MF, Sigaud M, Guerrero F, Neel A, et al. The factor VIII:C/VWF:Ag ratio as a useful tool to predict relapse in patients with acquired haemophilia A: A retrospective cohort study. Haemophilia 2019 ; 25 : 527-34
RISQUE ÉLEVÉ DE RÉCIDIVE À LONG TERME APRÈS UN PREMIER ÉPISODE DE THROMBOEMBOLIE VEINEUSE SURVENU PENDANT LA GROSSESSE OU EN POST-PARTUM : ÉTUDE REPEAT (RECURRENCE AFTER A PREGNANCY RELATED THROMBOSIS)
D’après : High risk of long-term recurrence after a first episode of venous thromboembolism during pregnancy or postpartum: the REcurrence after a PrEgnAncy related Thrombosis (REPEAT) Study. Ibrahim-Kosta M, El Harake S, Leclercq B, De Mari C, Secondi JF, Paoletti E, et al. J Thromb Haemost 2025 ; 23 : 937-46.
Actualité commentée réalisée par Laure MACRAIGNE
Justificatifs et objectifs
La maladie thromboembolique veineuse (MTEV) comprenant la thrombose veineuse (TVP) et sa complication principale, l’embolie pulmonaire (EP), fait partie des causes majeures de décès maternel en France et dans les pays développés.
Les facteurs de risque de thrombose liés à la grossesse sont bien établis, mais le risque global de récidive à long terme demeure encore mal caractérisé dans cette population spécifique.
L’objectif de l’étude REPEAT est de déterminer le taux de récidive tardive après un épisode de MTEV associé à une grossesse, et d’identifier plus précisément les facteurs de risque associés qui restent encore peu documentés à l’heure actuelle.
Méthodes
Entre 2000 et 2015, 587 patientes ayant présenté un premier épisode de MTEV lié à la grossesse ou au post-partum ont été incluses à l’hôpital de La Timone à Marseille. Les données cliniques et les résultats d’un bilan de thrombophilie ont été recueillis à l’inclusion. Parmi elles,
189 présentaient des antécédents de récidive de MTEV recueillis lors de consultations ultérieures. Un suivi réalisé entre 2016 et 2019 a permis de recontacter 230 des 398 patientes sans antécédents de récidive et non revenues en consultation, identifiant 32 nouveaux épisodes de
récidive. Ainsi au total, 221 épisodes de récidives de MTEV ont été observés chez les 587 patientes incluses.
Résultats
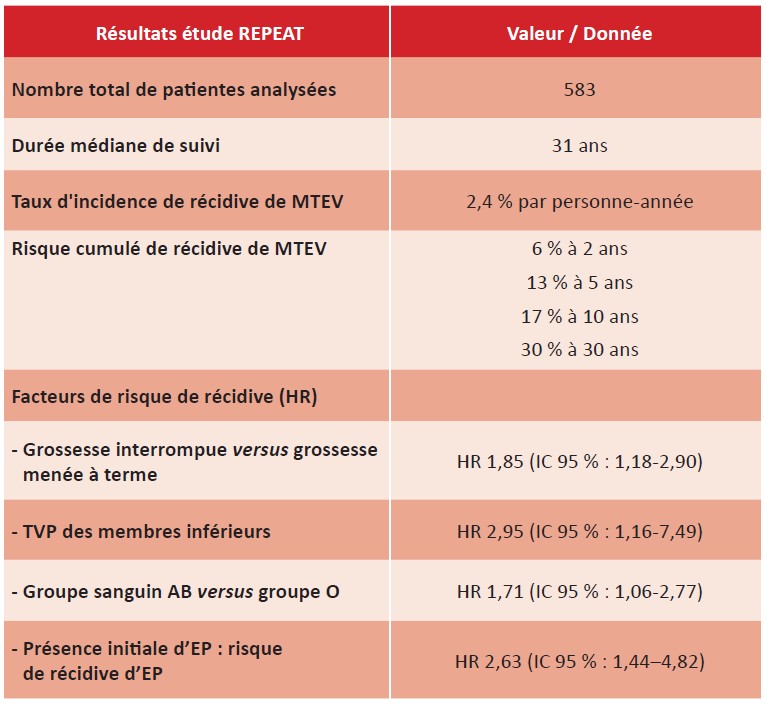
Sur 583 femmes analysées après contrôle qualité, le taux d’incidence de récidive de MTEV était de 2,4 % par personne année, avec un risque cumulé de 38 % à long terme (sur une médiane de suivi de 31 ans).
La présence initiale d’une EP doublait le risque de récidive sous forme d’EP. Les facteurs associés à la récidive étaient : grossesse interrompue, TVP des membres inférieurs et groupe sanguin AB.
Avis d’expert
Cette étude est la première à décrire le risque de récidive tardive, au-delà de 15 ans, d’une MTEV survenant pendant la grossesse ou le post-partum. Bien que le risque de récidive soit bien plus faible durant les 10 premières années que celui d’une MTEV non provoquée, plus d’un tiers des patientes présentent une récidive à 30 ans.
Cette étude montre que les patientes ayant présenté une EP lors du premier épisode ont un risque doublé de récidive sous forme d’EP par rapport à celles ayant eu une TVP initiale. Les facteurs de risque de récidive sont une grossesse non menée à terme, une thrombose des membres inférieurs (probablement liée à un syndrome post-thrombotique) et un groupe sanguin AB, reconnu comme facteur de risque génétique. En revanche, la thrombophilie biologique ne semble pas augmenter le risque de récidive.
Une limite importante réside dans l’absence d’informations systématiques sur la thromboprophylaxie, bien que plus de la moitié des récidives soient survenues dans des contextes à haut risque.
Ces résultats suggèrent qu’un suivi à très long terme est justifié chez les patientes ayant présenté une MTEV liée à la grossesse, même au-delà de dix ans. Les patientes présentant les facteurs de risque identifiés pourraient bénéficier d’une surveillance renforcée ou d’un traitement prophylactique prolongé.
L’AVENIR DES pARNi DANS LES APPROCHES THÉRAPEUTIQUES DE LA MALADIE DE VON WILLEBRAND
D’après : The future of siRNA-mediated approaches to treat von Willebrand disease. Linthorst NA, van Vlijmen BJM, Eikenboom JCJ. Expert Rev Hematol 2025 ; 18 : 109-22.
Actualité commentée réalisée par Geneviève MCCLUSKEY
Justificatifs et objectifs
La maladie de von Willebrand (VWD) est une maladie hémorragique caractérisée par un déficit qualitatif ou quantitatif en facteur Willebrand (VWF), et affecte entre 1/100 et 1/10 000 personnes. Les traitements pour la VWD ne sont pas optimaux, nécessitant des injections intraveineuses répétées, avec une efficacité parfois courte, aboutissant à une charge importante pour les patients. Aujourd’hui, il y a un développement important des thérapies à base d’ARN dans les maladies hémorragiques, en particulier les petits ARN interférents (pARNi). Les avantages sont nombreux : une efficacité à long terme et une guérison phénotypique. Cette revue propose de faire le point sur les avancées dans les traitements pARNi dans la VWD.
Résultats
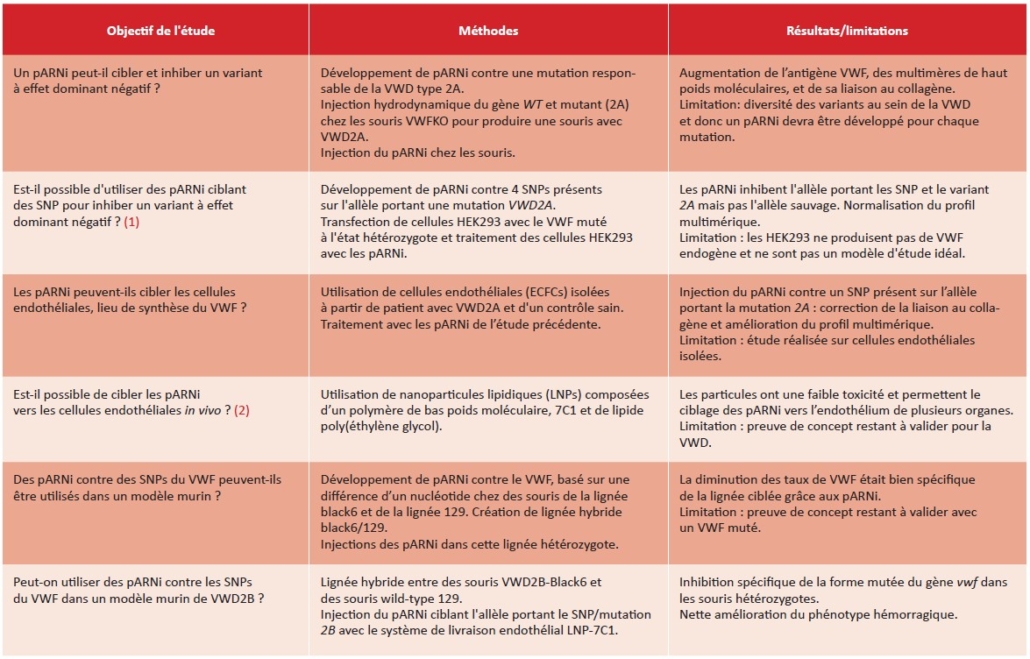
La revue présente les différentes études précliniques utilisant des pARNi dans la VWD. En particulier les auteurs illustrent l’efficacité et les avantages de cibler les « single nucleotide polymorphisms » (SNP) hétérozygotes dans le gène du vwf, en lieu et place de la mutation. Les SNPs sont en effet beaucoup plus fréquents dans la population, permettant de traiter plus de patients avec les mêmes outils, et font une distinction plus marquée entre les allèles, permettant de cibler spécifiquement la forme mutée et non l’allèle sain. Les différentes études réalisées in vitro ou dans des modèles murins sont présentées dans le tableau ci-dessous.
Avis d’expert
L’article « The Future of siRNA-Mediated Approaches to Treat von Willebrand Disease » présente les avantages significatifs de l’usage de pARNi dans la VWD. Les auteurs abordent les limites des traitements actuels, tels que les concentrés de facteur Willebrand ou la desmopressine, qui nécessitent des administrations fréquentes et qui ont des efficacités parfois limitées. Plus important encore, ces traitements n’éliminent pas les formes mutées et défectueuses du VWF, qui impactent le bon fonctionnement du VWF produit
par l’allèle sain. Les auteurs présentent très clairement les avancées précliniques des pARNi ciblant les SNPs hétérozygotes sur le gène du VWF pour inhiber spécifiquement l’allèle portant le variant. Globalement, cela permettrait de réduire sélectivement la production de VWF mutant tout en préservant l’expression de l’allèle sain. Cette approche permettrait une correction personnalisée du phénotype hémorragique à long terme. Les auteurs font référence à des données précliniques convaincantes, qui montrent des améliorations fonctionnelles du VWF après traitement par pARNi dans des modèles cellulaires ainsi que dans des modèles murins.
Cependant, plusieurs obstacles majeurs se présentent. Dans un premier temps, la livraison des pARNi dans les cellules endothéliales doit encore être optimisée. De plus, les pARNi sont rapidement dégradés dans la circulation, mais des technologies ont été développées pour diminuer cette dégradation (3). Par ailleurs, les études in vivo de ces pARNi sont difficilement transposables à l’homme étant donné les variations génétiques inter-espèces. Enfin, la variabilité génétique entre les patients complique l’application généralisée de ces thérapies. Néanmoins, les auteurs proposent que les modèles cellulaires établis, tels que les ECFS dérivées de patients, soient utilisés pour tester la sélectivité et l’efficacité des pARNi. Les modèles murins pouvant être, quant à eux, utilisés pour évaluer le profil de sécurité et la toxicité des pARNi.
En conclusion, malgré l’avenir prometteur des thérapies pARNi dans la maladie de Willebrand présenté dans cette revue, il reste encore des défis techniques et biologiques majeurs avant de passer à leur application clinique.
Références
1. De Jong A, Dirven RJ, Oud JA, Tio D, Van Vlijmen BJM, Eikenboom J.
Correction of a dominant-negative von Willebrand factor multimerization defect by small interfering RNA-mediated allele-specific inhibition of mutant von Willebrand factor. J Thromb Haemost 2018 ; 16 : 1357-68.
2. Dahlman JE, Barnes C, Khan OF, Thiriot A, Jhunjunwala S, Shaw TE, et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol 2014 ; 9 : 648-55.
3. Springer AD, Dowdy SF. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther 2018 ; 28 : 109-18