ÉDITION SPÉCIALE EAHAD 2026
19th Annual Congress of the European Association for Haemophilia and Allied Disorders
Du 3 au 6 février 2026, Dublin
Avec le soutien institutionnel
![]()
![]()
D’après la communication orale de A. Randi, Molecular mechanisms of angiodysplasia in VWD. 19th Annual Congress of the European Association of Haemophilia and Allied Disorders 2026 3-6 February
Antoine Babuty, Nantes
Le système vasculaire joue un rôle central dans l’hémostase, en interaction étroite avec les composants sanguins. Les angiodysplasies gastro-intestinales (GI), malformations vasculaires acquises touchant environ 0,8 % de la population générale, constituent la première cause de saignement digestif chez les sujets âgés. Le diagnostic est endoscopique et les analyses histologiques sont peu informatives, en faveur d’une architecture fragile et perméable. Les saignements GI sur angiodysplasie sont significativement plus fréquents chez les patients atteints de maladie de Willebrand (VWD) (5–20 %, jusqu’à 40 % en cas de VWD acquis sous assistance ventriculaire), particulièrement dans les formes avec perte des multimères de haut poids moléculaire (VWD types 2A, 2B, 3). Leur prise en charge est difficile et la réponse aux traitements, y compris anti-VEGF, reste inconstante. Plusieurs mécanismes physiopathologiques sont proposés dont l’augmentation tissulaire du VEGF et un déséquilibre de la balance angiopoïétine-2 (Ang2) / angiopoïétine-1 (Ang1). Le vieillissement contribuerait également aux angiodysplasies via l’inflammation chronique, la perte des péricytes et l’augmentation de la perméabilité vasculaire, en lien notamment avec le TNF-α et l’Ang2.
Le facteur Willebrand (VWF) apparaît comme un régulateur clé de l’homéostasie vasculaire. Il limite l’angiogenèse : son absence dans les cellules endothéliales augmente la migration et la formation de tubes vasculaires (Starke et. al, Blood 2011). Récemment, l’équipe de A. Randi a démontré que le VWF contrôle le stockage de l’Ang2 dans les corps de Weibel-Palade et régule sa synthèse via FOXO1. En l’absence de VWF et donc des corps de Weibel-Palade, l’Ang2 est libérée de façon excessive, et inhibe alors le récepteur Tie2 qui n’active plus la voie PI3K/AKT. FOXO1 non phosphorylé passe donc dans le noyau et favorise la transcription d’Ang2, entrainant une boucle d’amplification (Figure 1).
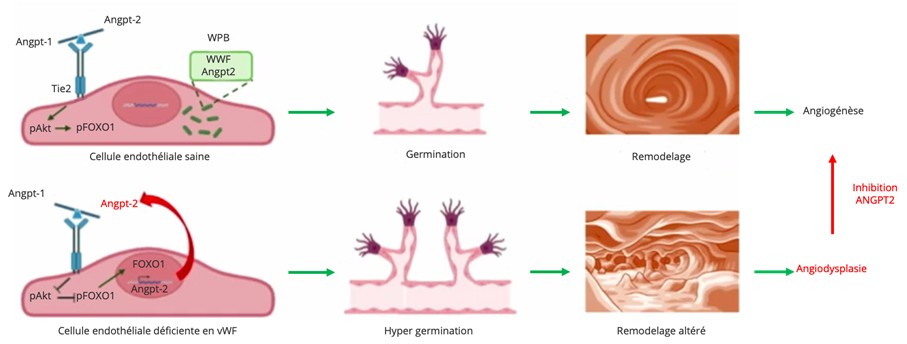
Figure 1 : Mécanismes moléculaires des angiodysplasies impliquant le facteur Willebrand et l’angiopoïétine 2
Ces résultats sont confirmés dans les modèles murins VWF-KO, qui présentent des anomalies marquées de la vascularisation intestinale et un déséquilibre de la balance Ang2/Ang1. Des modèles microfluidiques utilisant des cellules endothéliales issues de patients avec une VWD type 3 confirment une hyperangiogénèse de mauvaise qualité, réversible par blocage d’Ang2 (Figure 2) (Constantinescu-Bercu et al. Blood in press).
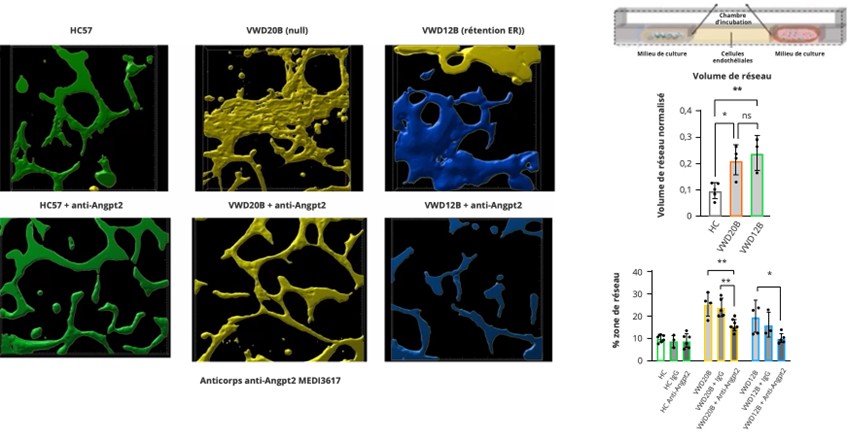
Figure 2 : L’inhibition de l’angiopoïétine 2 permet de normaliser l’angiogénèse in vitro à partir de cellules endothéliales issues de patients avec une VWD de type 3
A travers ces travaux remarquables, l’équipe de l’oratrice démontre, qu’au-delà des anomalies de l’hémostase, la VWD s’accompagne d’une dérégulation intrinsèque de l’angiogénèse, particulièrement marquée dans les formes sévères, et contribuant aux angiodysplasies et aux saignements récurrents digestifs et extra-digestifs. De plus, l’inhibition de l’angiopoiétine-2 semble une piste prometteuse dans le traitement des angiodysplasies GI chez les patients atteints de VWD.
Attention : ceci est un compte-rendu et/ou résumé des communications de congrès dont l’objectif est de fournir des informations sur l’état actuel de la recherche ; ainsi, les données présentées sont susceptibles de ne pas être validées par les autorités françaises et ne doivent donc pas être mises en pratique.
Ce compte-rendu a été réalisé sous la seule responsabilité du coordinateur, des auteurs et du directeur de la publication qui sont garants de l’objectivité de cette publication.