INTRODUCTION
L’anaphylaxie est la forme la plus sévère des réactions d’hypersensibilité immédiate, dont les symptômes peuvent aller d’une simple éruption cutanée à un état de choc anaphylactique, voire à l’apparition inaugurale d’un arrêt cardio-respiratoire. Les agents incriminés les plus courants sont les médicaments, les composants alimentaires et les piqûres d’hyménoptères. Il s’agit d’un événement rare dans la population générale (50-112 réactions par 100 000 personnes-année), dont l’incidence est néanmoins en constante augmentation (1). Certaines situations sont toutefois plus à risque, comme la période péri-opératoire avec une incidence de 1/353 à 1/18 600 anesthésies (2). La morbi-mortalité est alors plus élevée, estimée à 4,1 %, en cas de réaction à un curare, et ce, malgré un diagnostic rapide et un traitement adapté par remplissage vasculaire et administration d’adrénaline, qui constituent les traitements de référence (3). La physiopathologie de ces formes réfractaires à l’adrénaline n’est pas connue. Il est donc impératif d’améliorer notre compréhension des mécanismes moléculaires et cellulaires régissant l’anaphylaxie, en particulier les formes les plus graves. Des données récentes suggèrent que les plaquettes sanguines pourraient jouer un rôle central dans l’amplification de ces réactions. L’identification et la caractérisation du rôle des plaquettes dans l’anaphylaxie pourraient ouvrir de nouvelles perspectives diagnostiques et thérapeutiques afin d’améliorer la prise en charge des patients.
DÉFINITION
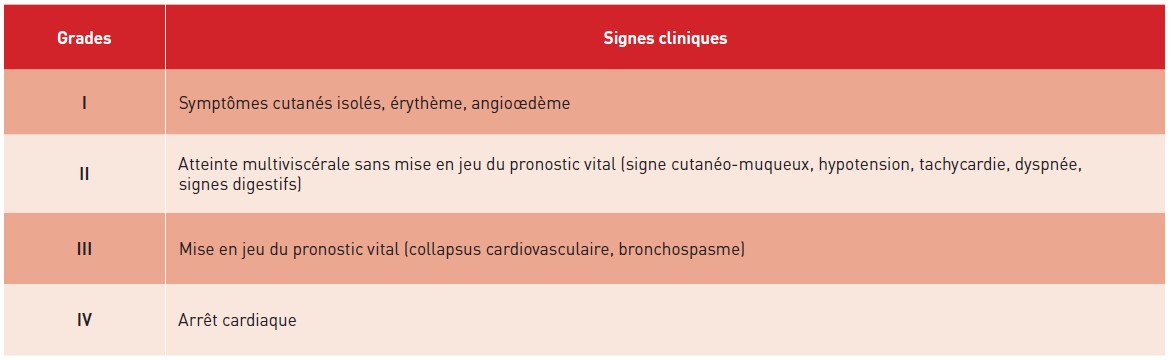
Le concept d’anaphylaxie a été découvert au début du XXe siècle par Charles Richet et Paul Portier, ce qui valut au premier le prix Nobel de physiologie en 1913. Lors de leurs études sur les toxines de méduses sur des cobayes, au lieu d’une immunisation attendue (phylaxie), une seconde injection provoqua une hypersensibilité mortelle, qu’ils nommèrent anaphylaxie. Ces travaux pionniers constituèrent le point de départ de la recherche sur l’anaphylaxie. Sur le plan clinique, l’anaphylaxie peut se manifester sous des formes variées, ce qui rend son diagnostic difficile, y compris pour des cliniciens expérimentés. Ainsi, la World Allergy Organization la définit comme étant une réaction d’hypersensibilité immédiate systémique et sévère, mettant potentiellement en jeu le pronostic vital. Cette définition ne présume pas du mécanisme de la réaction, qu’elle soit d’origine immunologique ou non. Les réactions anaphylactiques sont classées en quatre grades de sévérité croissante selon la classification de Ring et Messmer modifiée, permettant de guider la prise en charge thérapeutique (Tableau 1). Celle-ci comporte dans tous les cas l’éviction de l’allergène en cause, l’administration d’un remplissage vasculaire important ainsi que l’injection précoce d’adrénaline (de 10 à 1000 μg par voie intraveineuse, selon la sévérité de la réaction) (3). D’autres traitements peuvent également être mis en place comme une corticothérapie pour limiter la durée de l’inflammation ou des β2-mimétiques qui induisent la relaxation des muscles lisses bronchiques en cas de bronchospasme.
Malgré une prise en charge rapide par des anesthésistes réanimateurs, la morbidité et mortalité restent significatives, notamment dans des situations à haut risque comme la période péri-opératoire. L’existence de ces formes mortelles, malgré une réanimation bien conduite, suggère l’existence de chocs anaphylactiques réfractaires au traitement par adrénaline. La physiopathologie de ces formes spécifiques n’est pour l’heure pas connue mais pourrait comporter des boucles d’amplification de la réaction, impliquant l’activation secondaire d’autres éléments cellulaires.
MÉCANISMES
Plusieurs mécanismes peuvent être responsables d’une réaction anaphylactique, certains mettant en oeuvre l’immunité adaptative (réaction médiée par les IgE ou les IgG), tandis que d’autres sont indépendants comme la libération d’histamine non-spécifique, l’activation mastocytaire directe via le récepteur MRGPRX-2, l’activation de la phase contact ou du système kinine-kallicréine.
La réaction médiée par les immunoglobulines de type E (IgE) constitue la réaction d’hypersensibilité immédiate la plus connue et la mieux décrite, classée comme la réaction d’hypersensibilité de type I décrite par Gell et Coombs, et comprend deux phases. Une première phase de sensibilisation, qui fait suite à un premier contact avec l’allergène, mobilise les cellules présentatrices d’antigène qui le capturent, l’internalisent et exposent un de ses fragments à leur surface, lié au complexe majeur d’histocompatibilité (CMH) de classe II. Cette association antigène/CMH de classe II est reconnue par les lymphocytes T, qui se polarisent alors vers un profil Th2. Ces Th2 sécrètent des cytokines (IL-4, IL-13) favorisant l’activation des lymphocytes B, qui se différencient en plasmocytes, subissent une commutation isotypique et produisent des IgE spécifiques de l’allergène. Les IgE se fixent par la suite sur leurs récepteurs de haute affinité FcεRI, présents sur les mastocytes et basophiles. Cette phase dure une dizaine de jours et reste silencieuse d’un point de vue des signes cliniques. Lorsque le patient est à nouveau exposé au même allergène, ce dernier se fixe sur les IgE spécifiques, déjà présentes sur les cellules effectrices (mastocytes et basophiles), ce qui entraîne une dimérisation des récepteurs FcεRI conduisant alors à la transduction du signal. Ceci provoque l’activation de ces cellules effectrices conduisant à une libération massive de médiateurs préformés présents dans leurs granules (histamine, tryptase), ainsi qu’une synthèse et une sécrétion rapide de médiateurs néoformés d’origine lipidique (leucotriènes, prostaglandines, Platelet Activating Factor -PAF). Cette phase effectrice de l’allergie est à l’origine du phénotype clinique de l’anaphylaxie. Cependant, la voie médiée par les IgE est insuffisante pour expliquer l’ensemble des réactions anaphylactiques observées en pratique clinique, comme le montre l’absence d’identification de l’allergène mis en cause dans de nombreux cas, malgré un bilan exhaustif chez des patients ayant enduré une véritable réaction anaphylactique. Cela constitue un réel problème en rendant impossible un conseil sûr et expose le patient à un risque de récurrence de la réaction.
L’hypothèse d’un mécanisme impliquant la production d’immunoglobulines de type G (IgG) dirigées contre l’allergène, expliquant alors l’absence d’IgE spécifiques, a ainsi été proposée. Ce mécanisme correspond à l’hypersensibilité de type 3 dans la classification de Gell et Coombs. Les IgG spécifiques, produites lors de la phase de sensibilisation, reconnaissent leur antigène et forment des complexes immuns qui interagissent avec leurs récepteurs spécifiques de la famille FcγR. Ce mécanisme a formellement été identifié dans des modèles murins en faisant intervenir les récepteurs FcγRIII et FcγRIV (4). Chez l’homme, c’est le récepteur FcγRIIA, aussi appelé CD32a, qui pourrait jouer un rôle prépondérant (5). Il est présent à la surface de nombreuses cellules, notamment les neutrophiles, les basophiles et les plaquettes sanguines. Ces éléments cellulaires seraient les principaux effecteurs des réactions médiées par les IgG avec une sécrétion particulièrement importante de myélopéroxydase, sécrétée par les neutrophiles, ainsi que le Platelet Activating Factor (PAF) issu des neutrophiles et des plaquettes, et la sérotonine libérée par les plaquettes activées. Le PAF est un médiateur lipidique d’intérêt majeur, car il induit une vasodilatation, augmente la perméabilité des vaisseaux, stimule l’activation des mastocytes (notamment pulmonaires) et déclenche l’agrégation plaquettaire. Il existe une corrélation entre la sévérité de la réaction et la concentration plasmatique de PAF, faisant de ce médiateur une cible thérapeutique prometteuse. L’évolution vers un état de choc réfractaire au traitement par adrénaline est encore mal comprise et pourrait être liée à l’activation secondaire d’autres éléments cellulaires comme les plaquettes.
LES PLAQUETTES : NOUVEL ACTEUR DE L’ANAPHYLAXIE
Le rôle des plaquettes dans l’anaphylaxie est suspecté depuis les années 1970 grâce à l’utilisation de modèles d’anaphylaxie active chez le lapin (6), mais aussi le cochon d’Inde et la souris. Ces travaux mettent en évidence l’apparition brutale et transitoire d’une thrombopénie parfois associée à une neutropénie avec séquestration pulmonaire tandis que l’induction d’une thrombopénie préalablement à l’induction du choc en atténue la sévérité (6). L’idée a longtemps prévalu que les plaquettes possédaient le récepteur FcεRI, les désignant comme des effecteurs primaires de l’anaphylaxie médiée par les IgE ; cette hypothèse a cependant été invalidée par la preuve récente que les plaquettes humaines n’expriment ni les récepteurs FcεRI, ni les récepteurs FcεRII (7). Les plaquettes peuvent cependant être activées indirectement via divers médiateurs secondaires comme le PAF libéré par les basophiles et mastocytes ou encore la thrombine produite lors de l’activation de la phase contact de la coagulation, déclenchée par l’héparine et les polyphosphates sécrétés par les mastocytes.
En revanche, les plaquettes humaines sont directement sensibles aux IgG car elles possèdent à leur surface un récepteur aux IgG, le récepteur FcγRIIA (CD32a), tout comme les neutrophiles et les basophiles. Ce récepteur possède une forte affinité pour les complexes immuns d’IgG, mais une faible affinité pour les IgG seules. Le rôle de FcγRIIA dans l’anaphylaxie médiée par les IgG a récemment été décrit à l’aide de souris transgéniques humanisées pour le récepteur FcγRIIA, les souris de type « sauvage » étant naturellement dépourvues de ce récepteur (8). L’injection d’IgG agrégées par la chaleur provoque une diminution de la température corporelle, constituant ainsi un marqueur indirect d’anaphylaxie. Une thrombopénie précoce est également observée, mais uniquement chez les souris humanisées. De plus, la déplétion des plaquettes effectuée avant l’induction du choc supprime complètement la réaction. À l’inverse, une thrombocytose, induite par l’injection de romiplostime, analogue de la thrombopoïétine, conduit à l’aggravation de la réaction. Celle-ci est associée à la libération massive de sérotonine, présente dans les granules plaquettaires. L’élimination de la sérotonine plaquettaire ou l’injection d’un antagoniste des récepteurs de la sérotonine réduit alors la sévérité de la réaction dans ce modèle.
La transposition de ces résultats chez l’homme est pour l’instant limitée. Une étude prospective multicentrique menée chez 86 patients suspectés de choc anaphylactique per-opératoire à un curare myorelaxant (étude « NASA » Neutrophil Activation in Systemic Anaphylaxis) a rapporté une diminution du nombre de plaquettes circulantes dans les cas d’anaphylaxie grave, associée à la présence de plaquettes activées (9). Cependant, le protocole de cette étude n’était pas optimisé pour l’analyse des plaquettes, soulignant la nécessité de mener d’autres études cliniques pour confirmer ces observations. De plus, l’existence même d’une voie IgG dans l’anaphylaxie humaine reste matière à débat en raison du manque d’outils permettant de différencier ces réactions de celles médiées par les IgE. Enfin, l’hypothèse d’une implication des deux voies IgE et IgG semble plausible aussi, en particulier dans les formes les plus graves (9).
RÔLES POTENTIELS DES PLAQUETTES DANS LA SÉVÉRITÉ DES RÉACTIONS ANAPHYLACTIQUES
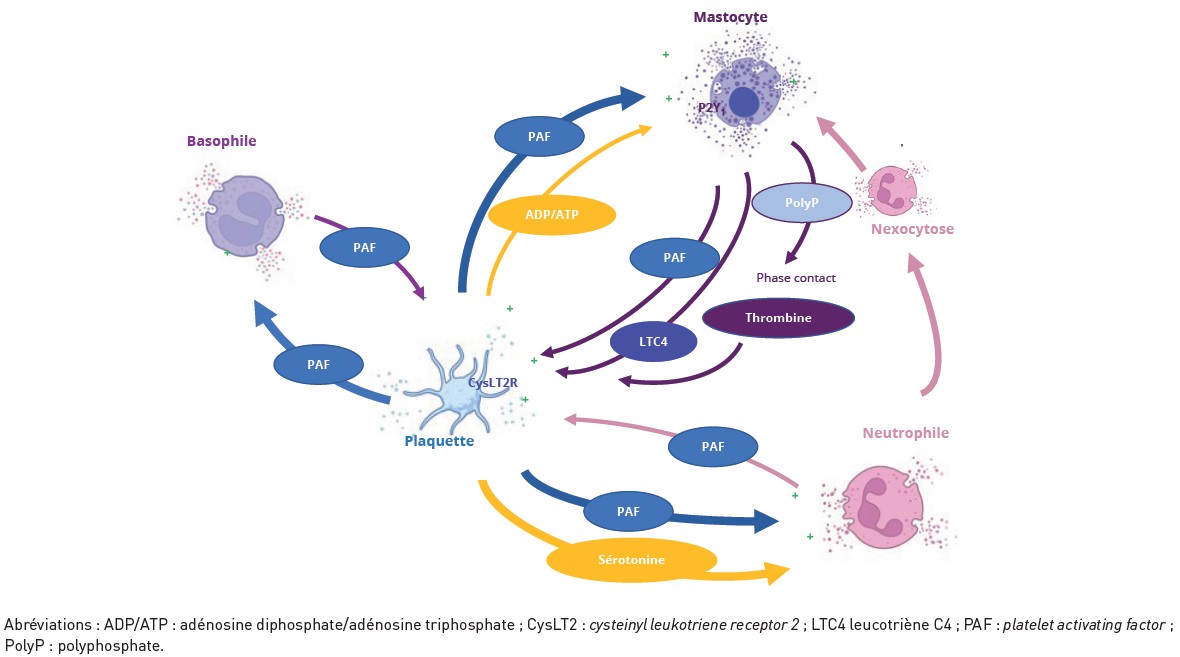
Les plaquettes peuvent contribuer de manières diverses à la sévérité de la réaction et différentes hypothèses peuvent être proposées en s’appuyant sur les données récentes de la littérature. Comme évoqué plus haut, les plaquettes pourraient jouer un rôle dans l’amplification de la réaction anaphylactique en libérant leur contenu granulaire riche en différents médiateurs, comme le PAF et la sérotonine. En effet, ces deux médiateurs exercent des effets cardiovasculaires et respiratoires compatibles avec ceux observés au cours de l’anaphylaxie (Figure 1). Peu d’études se sont intéressées au rôle de la sérotonine au cours de l’anaphylaxie. Une seule étude a montré que la sérotonine, principalement stockée dans les granules denses des plaquettes, est libérée lorsque le sang de patients ayant présenté un choc anaphylactique aux curares est exposé in vitro à ces agents (10). Ces résultats suggèrent que la sérotonine pourrait participer à l’aggravation des manifestations cliniques. Toutefois, cette étude a été menée uniquement in vitro, sur sang total, et portait sur un effectif limité de dix patients. Outre les plaquettes, il a également été démontré une activation directe des neutrophiles par la sérotonine (10).
Les plaquettes pourraient également établir des communications avec les mastocytes par l’intermédiaire de médiateurs et ainsi influencer leur phénotype. Le PAF apparaît comme un médiateur central dans les interactions entre plaquettes et mastocytes, ces derniers pouvant être activés par le PAF libéré par les plaquettes lors de situations inflammatoires (circulation extracorporelle, anaphylaxie passive) (11). La libération du PAF contenu dans les plaquettes pourrait ainsi aggraver la symptomatologie clinique, à la fois par l’action directe du PAF sur les tissus cibles (endothélium et muscles lisses vasculaires, muscles lisses bronchiques) mais aussi en amplifiant l’activation mastocytaire. Il a été par ailleurs montré que les plaquettes peuvent activer directement les mastocytes pulmonaires en culture en sécrétant du PAF (11). Cette action pourrait contribuer à la sévérité de la réaction au moyen de l’activation des mastocytes à distance du site d’inoculation de l’allergène, propageant ainsi la réponse anaphylactique. En outre, les plaquettes pourraient également être activées secondairement. En effet, des travaux récents montrent que l’activation des mastocytes par l’IL-33, associée à la sécrétion de LTC4, initie une activation des plaquettes via les récepteurs CysLT2 et induit la libération de nucléotides (ADP/ATP). Ces nucléotides stimulent ensuite le récepteur P2Y1 des mastocytes, ce qui amplifie la production de LTC4 et favorise la libération de médiateurs allergiques, notamment la PGD2 et l’histamine (13).
Par ailleurs, des études récentes montrent que les mastocytes périvasculaires peuvent interagir directement avec les cellules circulantes grâce à leurs protubérances intraluminales (12) et qu’après activation, les mastocytes piègent les neutrophiles au sein de leurs vacuoles intracellulaires, puis recyclent leurs composants afin de renforcer leur propre activité inflammatoire par l’exocytose de molécules provenant des neutrophiles (nexocytose) (13). L’implication des plaquettes dans ce mécanisme n’est pour l’heure pas établie, mais ne saurait être exclue compte tenu des interactions étroites qui peuvent s’établir entre plaquettes et neutrophiles, susceptibles de favoriser leur internalisation par les mastocytes.
CONCLUSION
Un faisceau d’arguments expérimentaux concordants suggère l’implication des plaquettes dans le choc anaphylactique chez la souris et leur contribution à la sévérité de la réaction. Toutefois, les mécanismes sousjacents nécessitent encore d’être approfondis. De même, les preuves de l’implication des plaquettes dans l’anaphylaxie chez l’homme sont encore manquantes. Des études cliniques prospectives menées notamment dans le contexte péri-opératoire, bien contrôlé, devraient pouvoir apporter des réponses. Ces données pourraient ouvrir la voie à l’utilisation de nouveaux outils diagnostiques pour caractériser les mécanismes à l’origine de la réaction, mais aussi à l’utilisation de nouveaux agents thérapeutiques pour prendre en charge les patients les plus graves, ayant un état de choc anaphylactique réfractaire au traitement de référence, l’adrénaline.
POINTS CLÉS À RETENIR
• L’anaphylaxie est la forme la plus grave des réactions d’hypersensibilité dont le traitement de référence, l’adrénaline, est parfois mis en défaut, à l’origine d’une mortalité significative.
• Les réactions médiées par les IgE sont insuffisantes pour expliquer l’ensemble des réactions anaphylactiques.
• Une voie alternative de l’anaphylaxie pourrait être médiée par les IgG en faisant intervenir les neutrophiles et les plaquettes sanguines.
• Les plaquettes sanguines pourraient jouer un rôle dans l’amplification de la réaction anaphylactique par la libération des médiateurs PAF et sérotonine, qui constituent des cibles thérapeutiques potentielles.
Liens d’intérêts : Charles-Ambroise TACQUARD déclare un essai clinique : étude HEMOCANOPE ; les autres auteurs déclarent ne pas avoir de lien d’intérêt en rapport avec cet article.